UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark one)
ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______ to _______.
Commission file number
(Exact name of registrant as specified in its charter)
| ||
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
|
| |
| ||
(Address of principal executive offices) | (Zip Code) |
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol |
| Name of each exchange on which registered |
| The |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ☐ |
| |
Non-accelerated filer ☐ | Smaller reporting company | |
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of June 30, 2021 was $
As of March 8, 2022,
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement for the registrant’s 2022 annual meeting of stockholders to be filed within 120 days after the end of the period covered by this Annual Report on Form 10-K are incorporated by reference into Part III of this Annual Report on Form 10-K.
ANI PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2021
TABLE OF CONTENTS
|
|
| Page |
|
|
| |
| 3 | ||
| 15 | ||
| 38 | ||
| 39 | ||
| 39 | ||
| 39 | ||
|
|
| |
|
| ||
| 39 | ||
Reserved |
| 42 | |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
| 43 | |
| 63 | ||
| 65 | ||
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
| 110 | |
| 110 | ||
| 111 | ||
Item 9C. | Disclosures Regarding Foreign Jurisdictions that Prevent Inspections | 99 | |
|
|
| |
|
| ||
| 111 | ||
| 112 | ||
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
| 112 | |
Certain Relationships and Related Transactions, and Director Independence |
| 112 | |
| 112 | ||
|
|
| |
|
| ||
| 112 | ||
| 118 | ||
|
| ||
| 119 | ||
In this annual report, references to “ANI Pharmaceuticals,” “ANI,” the “Company,” “we,” “us,” and “our” refer, unless the context requires otherwise, to ANI Pharmaceuticals, Inc., a Delaware corporation, and its consolidated subsidiaries. References to “named executive officers” refer to our current named executive officers, except where the context requires otherwise.
CAUTIONARY STATEMENT CONCERNING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and certain information incorporated herein by reference contain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Such statements include, but are not limited to, the announcement of the acquisition of Novitium Pharma LLC (“Novitium”), statements about future operations, strategies and growth potential, the revenue potential (licensing, royalty and sales) of products we sell, development timelines, expected timeframe for submission of new drug applications or supplemental new drug applications to the U.S. Food and Drug Administration (the “FDA”), pipeline or potential markets for our products, selling and marketing strategies and associated costs to support the sales of Purified Cortrophin™ Gel (Repository Corticotropin Injection USP) (“Cortrophin Gel”), impact of accounting principles, litigation expenses, liquidity and capital resources, the impact of the novel coronavirus (“COVID-19”) global pandemic on our business, and other statements that are not historical in nature, particularly those that utilize terminology such as “anticipates,” “will,” “expects,” “plans,” “potential,” “future,” “believes,” “intends,” “continue,” other words of similar meaning, derivations of such words, and the use of future dates. Such forward-looking statements are based on the reasonable beliefs of our management as well as assumptions made by and information currently available to our management. Readers should not put undue reliance on these forward-looking statements. Forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified; therefore, our actual results may differ materially from those described in any forward-looking statements. Factors that might cause such a difference include, but are not limited to, those discussed in our periodic reports filed with the U.S. Securities and Exchange Commission (the “SEC”), including those discussed in the “Risk Factors” section in Part I, Item 1A.of this Annual Report on Form 10-K, and the following factors:
| • | risks that we may face with respect to importing raw materials; |
| • | delays or failure in obtaining approvals by the FDA of the products we sell; |
| • | changes in policy or actions that may be taken by the FDA and other regulatory agencies, including drug recalls; |
| • | the ability of our manufacturing partners to meet our product demands and timelines; |
| • | our dependence on single source suppliers of ingredients due to the time and cost to validate a second source of supply; |
| • | acceptance of our products at levels that will allow us to achieve profitability; |
| • | our ability to develop, license or acquire, and commercialize new products; |
| • | the level of competition we face and the legal, regulatory and/or legislative strategies employed by our competitors to prevent or delay competition from generic alternatives to branded products; |
| • | our ability to protect our intellectual property rights; |
| • | the impact of legislative or regulatory reform on the pricing for pharmaceutical products; |
| • | the impact of any litigation to which we are, or may become, a party; |
| • | our ability, and that of our suppliers, development partners, and manufacturing partners, to comply with laws, regulations and standards that govern or affect the pharmaceutical and biotechnology industries; |
1
| • | our ability to maintain the services of our key executives and other personnel; and |
| • | general business and economic conditions and the effects and duration of outbreaks of public health emergencies, such as COVID-19. |
NOTE REGARDING TRADEMARKS
Apexicon®, Cortenema®, Purified Cortrophin™ Gel, Cortrophin-Zinc®, Inderal® LA, Inderal® XL, InnoPran XL®, Lithobid®, Reglan®, Vancocin®, and Veregen® are registered trademarks subject to trademark protection and are owned by ANI Pharmaceuticals, Inc. and its consolidated subsidiaries. Atacand® and Atacand HCT® are the property of AstraZeneca AB and are licensed to ANI Pharmaceuticals, Inc. for U.S. sales of those products. Arimidex® and Casodex® are the property of AstraZeneca UK Limited and are licensed to ANI Pharmaceuticals, Inc. for U.S. sales of those products. Oxistat® is the property of Fougera Pharmaceuticals Inc. and licensed to ANI Pharmaceuticals, Inc. for U.S. sales of Oxistat® Lotion. Pandel® is property of Taisho Pharmaceutical Co, Ltd. and licensed to ANI Pharmaceuticals for U.S. sales of Pandel® creme.
2
PART I
Item 1. Business
ANI Pharmaceuticals, Inc. and its consolidated subsidiaries (together, “ANI,” the “Company,” “we,” “us,” or “our”) is a diversified bio-pharmaceutical company serving patients in need by developing, manufacturing, and marketing high quality branded and generic prescription pharmaceuticals, including for diseases with high unmet medical need. Our team is focused on delivering sustainable growth by building a successful Purified Cortrophin Gel franchise, strengthening our generics business with enhanced development capability, innovation in established brands and leveraging our North American manufacturing capabilities. Our four pharmaceutical manufacturing facilities, of which two are located in Baudette, Minnesota, one is located in East Windsor, New Jersey, and one is located in Oakville, Ontario, are together capable of producing oral solid dose products, as well as semi-solids, liquids and topicals, controlled substances, and potent products that must be manufactured in a fully-contained environment.
Through research and development, acquisitions of businesses, acquisitions of Abbreviated New Drug Applications (“ANDAs”), New Drug Applications (“NDAs”), product rights, and entry into agreements to obtain the distribution rights for various products, we have a commercial portfolio of 101 products with a wide variety of indications and a robust portfolio of pipeline products as of December 31, 2021. Refer to our website at www.anipharmaceuticals.com for information on our products, including indications/treatments.
On August 6, 2018, our subsidiary, ANI Pharmaceuticals Canada Inc. (“ANI Canada”), acquired all the issued and outstanding equity interests of WellSpring Pharma Services Inc. (“WellSpring”), a Canadian company that performs contract development and manufacturing of pharmaceutical products. In conjunction with the transaction, we acquired WellSpring’s pharmaceutical manufacturing facility, laboratory, and offices, its current book of commercial business, as well as an organized workforce.
On November 19, 2021, we completed the acquisition of Novitium Pharma LLC (“Novitium”). With operations in East Windsor, New Jersey, and Chennai, India, Novitium is a pharmaceutical company that specializes in development, manufacturing, and distribution of niche generic products. Founded in 2016, Novitium has since developed a growing commercial product portfolio spanning a diverse range of dosage forms and therapeutic categories.
Unless otherwise required by the context, references in this Annual Report on Form 10-K to the “Company,” “we,” “us,” and “our” refer to ANI Pharmaceuticals, Inc., a Delaware corporation formed in April 2001. Our principal executive offices are located at 210 Main Street West, Baudette, Minnesota, 56623, our telephone number is (218) 634-3500, and our website address is www.anipharmaceuticals.com.
Strategy
Our objective is to build a sustainable and growth oriented biopharmaceutical company serving patients in need and creating long-term value for our investors. Our growth strategy is driven by the following key pillars:
Building a successful Cortrophin Gel franchise.
We acquired the NDAs for Cortrophin gel and Cortrophin-Zinc in January 2016 and executed long-term supply agreements with a supplier of our primary raw material for corticotrophin active pharmaceutical ingredient (“API”), a supplier of corticotrophin API with whom we have advanced the manufacture of commercial scale batches of API, and a Cortrophin gel fill/finish contract manufacturer. During the second quarter of 2021, we submitted a Supplemental New Drug Application (“sNDA”) to the FDA.
On October 29, 2021, the FDA approved the Company’s sNDA for Purified Cortrophin™ Gel (Repository Corticotropin Injection USP) for the treatment of certain chronic autoimmune disorders, including acute exacerbations of multiple sclerosis (“MS”) and rheumatoid arthritis (“RA”), in addition to excess urinary protein due to nephrotic syndrome. Cortrophin Gel is an adrenocorticotropic hormone (“ACTH”), also known as purified corticotropin.
3
During 2021, we invested in leadership, expertise and infrastructure in the areas of commercialization of rare disease therapies and developed a launch strategy and commercial plan for this product. In the fourth quarter of 2021 and first quarter of 2022, we hired a significant number of new employees and assembled and trained our rare disease field force. On January 24, 2022, we announced the commercial launch of Cortrophin Gel in the U.S. As a result of the build out of our rare disease team, our expenditures in support of these efforts are expected to materially increase in 2022 as compared to 2021.
Strengthening our generics business with enhanced research and development capability and increased focus on niche opportunities
We have grown our generics business through a combination of market share gains on existing products and new product launches. We have also successfully acquired numerous ANDAs through business and asset acquisitions, including, most recently, our acquisition of Novitium, including their portfolio of commercial and pipeline generic products, manufacturing and development facilities and expert workforce. We have begun to increase our focus on niche lower competition opportunities such as injectables, Paragraph IV, and Competitive Generic Therapy designation filings. Additionally, we will continue to seek opportunities to enhance our capabilities through strategic partnerships and acquisitions of assets and businesses.
Maximizing the value from our established brands through innovative “go-to-market” (“GTM”) strategies and continued programmatic acquisitions
We have acquired the New Drug Applications (“NDAs”) for and market Atacand, Atacand HCT, Arimidex, Casodex, Lithobid, Vancocin, Inderal LA, Inderal XL, InnoPran XL, Oxistat, Veregen, and Pandel. We are innovating in our GTM strategy through creative partnerships. In addition, we will continue to explore opportunities in acquiring new brands to grow our established brands portfolio.
Expansion of contract development and manufacturing organization (“CDMO”) business by leveraging our unique manufacturing capabilities
We built a CDMO business through our sites in Baudette and grew it through the acquisition of Novitium and WellSpring. Our North America based manufacturing and unique capabilities in high-potency, hormonal, steroid, and oncolytic products can be leveraged to expand our CDMO business.
The pillars of our strategy are enabled by an empowered, collaborative, and purposeful team with a high performance-orientation.
Product Development Considerations
We consider a variety of criteria in determining which products to develop, all of which influence the level of competition upon product launch. These criteria include:
| ● | Formulation Complexity. Our development and manufacturing capabilities enable us to manufacture pharmaceuticals that are difficult to produce, including highly potent, extended release, combination, and low dosage products. This ability to manufacture a variety of complex products is a competitive strength that we intend to leverage in selecting products to develop or manufacture. |
| ● | Patent Status. We seek to develop products whose branded bioequivalents do not have long-term patent protection or existing patent challenges. |
| ● | Market Size. When determining whether to develop or acquire an individual product, we review the current and expected market size for that product at launch, as well as forecasted price erosion upon conversion from branded to generic pricing. We endeavor to manufacture products with sufficient market size to enable us to enter the market with a strong likelihood of being able to price our products both competitively and at a profit. |
4
| ● | Profit Potential. We research the availability and cost of active pharmaceutical ingredients in determining which products to develop or acquire. In determining the potential profit of a product, we forecast our anticipated market share, pricing, including the expected price erosion caused by competition from other generic manufacturers, and the estimated cost to manufacture the products. |
| ● | Manufacturing. We generally seek to develop and manufacture products at our own manufacturing plants in order to optimize the utilization of our facilities, ensure quality control in our products, and to more closely control the economic inputs and outputs of our products. |
| ● | Competition. When determining whether to develop or acquire a product, we research existing and expected competition. We seek to develop products for which we can obtain sufficient market share and may decline to develop a product if we anticipate significant competition. Our specialized manufacturing facilities provide a means of entering niche markets, such as hormone therapies, in which fewer generic companies are able to compete. |
In addition to laboratories that support the requirements of raw material, finished product, and stability testing, we have a 1,000-square foot pilot laboratory offering liquid, suspension and solid dose development capabilities. This pilot laboratory offers a full range of analytical capabilities, including method development, validation and de-formulation, and is licensed by the Drug Enforcement Administration (“DEA”). Finally, a separate development suite located within our high-potency manufacturing facility offers additional capabilities for product development.
Products and Markets
Products
A complete list of our generic and branded pharmaceutical products and descriptions is posted on our website, www.anipharmaceuticals.com.
In November 2021, we acquired Novitium which specializes in development, manufacturing, and distribution of niche generic products. At the time of the acquisition, Novitium’s commercial portfolio consisted of 24 generic products.
Markets
In determining which products to pursue for development, we target products that are complex to manufacture and therefore have higher barriers to entry. These factors provide opportunities for growth, utilizing our competitive strengths at the same time that they decrease the number of potential competitors in the markets for these products. These markets currently include controlled substances, oncology products, hormones and steroids, injectables, and complex formulations, including extended release and combination products.
Controlled Substances
Schedule II controlled substances are drugs considered to have a high abuse risk but that also have safe and accepted medical uses. In addition to our Schedule II products currently on the market, our pipeline includes ANDAs in this market. One of our manufacturing facilities in Baudette, Minnesota and our manufacturing facility in East Windsor, New Jersey is licensed by the DEA for the manufacture of Schedule II controlled substances. Our manufacturing facility in Oakville, Ontario is licensed by Health Canada for the manufacture of Schedule II controlled substances.
Oncology Products
Due to the capabilities of our containment facility and our expertise in manufacturing segregation, we are focused on developing and manufacturing niche oncology products (anti-cancer). In particular, we are targeting products subject to priority review by the FDA, more specifically those with no blocking patents and no generic competition. We currently have a variety of oncology products on the market.
5
Hormone and Steroid Drugs
The market for hormone and steroid drugs includes hormone therapy to alleviate menopausal symptoms in women, contraceptives, testosterone replacement therapies for men, and therapies for treating hormone-sensitive cancers.
Hormone Therapy (“HT”) has been a long-accepted medical treatment for alleviating the symptoms of menopause. Initially, HT consisted of estrogen only but has evolved to include combination therapies of estrogen, progesterone, and androgens. We target niche products in the HT and steroid product market for several reasons, including:
| ● | Hormone and steroid products are a core competency based on our manufacturing and product development teams’ long history of manufacturing these types of products; and |
| ● | The aging “baby boomer” population, of which women represent a majority, is expected to support continued growth in the HT market. |
Injectables
Our burgeoning injectable portfolio contains injectable ANDA products encompassing several key therapeutic areas. Cortrophin Gel is our first branded injectable product. We work with world-class manufacturing partners to support these efforts.
Complex Formulations
We have a range of complex formulation products currently on the market and a pipeline that includes various extended-release products and combination products.
Competitive Generic Therapy
The FDA Reauthorization Act of 2017, or (“FDARA”), created a new pathway by which FDA may, at the request of the applicant, designate a drug with “inadequate generic competition” as a competitive generic therapy (“CGT”). At the request of the applicant, the FDA may also expedite the review of an ANDA for a drug designated as a CGT. Under the CGT pathway, the FDA provides a statutory provision for a 180-day exclusivity period for certain first to market applicants whose ANDA received a CGT designation. Our Novitium subsidiary has developed a strong track record of obtaining CGT approvals and we expect to continue to develop generic drugs under the CGT pathway.
Contract Manufacturing
We manufacture pharmaceutical products for several branded and generic companies, who outsource production in order to:
| ● | Free-up internal resources to focus on sales and marketing as well as research and development; |
| ● | Employ internal capacity to manufacture higher volume or more critical products; and |
| ● | Utilize our specialized equipment and expertise. |
Given our specialized manufacturing capabilities, we are focused on attracting niche contract manufacturing opportunities that offer high margins.
In conjunction with our acquisitions of WellSpring and Novitium, we acquired WellSpring’s and Novitium’s pharmaceutical manufacturing facilities. As a result of these transactions, we perform contract manufacturing in our Baudette, Minnesota, Oakville, Ontario, and East Windsor, New Jersey facilities.
6
Manufacturing, Suppliers, and Raw Materials
We require a supply of quality raw materials, including active pharmaceutical ingredients (“API”), and components to manufacture and package our pharmaceutical products. In order to manufacture certain of our products deemed controlled substances, we must submit a request to the DEA for a quota to purchase the amount of API needed for manufacture. Without approved quotas from the DEA, we would not be able to purchase these ingredients from our suppliers.
We source the raw materials for our products from both domestic and international suppliers, which we carefully select. Generally, we qualify only a single source of API for use in each product due to the cost and time required to validate and qualify a second source of supply. Any change in one of our API suppliers must usually be approved through a Prior Approval Supplement (“PAS”) by the FDA. The process of obtaining an approval of such a PAS can require between four and 18 months. While we also generally qualify a single source for non-API raw materials, the process required to qualify an alternative source of a non-API raw material is typically much less rigorous. If we were to change the supplier of a raw material for a product, the cost for the material could be greater than the amount we paid with the previous supplier. Changes in suppliers are rare but could occur as a result of a supplier’s business failing, an issue arising from an FDA inspection, or failure to maintain our required standards of quality. As a result, we selectively choose suppliers based on various factors including quality, reliability of supply, and long-term financial stability.
Certain of the APIs for our drug products, including those that are marketed without approved NDAs or ANDAs, are sourced from international suppliers. From time to time, we have experienced temporary disruptions in the supply of certain of such imported API due to FDA inspections and customs delays. In addition, certain of our products are manufactured, packaged, or manufactured and packaged by third parties.
Government Regulation
The pharmaceutical industry in the U.S. and Canada is highly regulated by multiple U.S. and Canadian government agencies, such as the FDA, the DEA, the Centers for Medicare and Medicaid Services (“CMS”), and Health Canada. As a result, we are subject to extensive and complex rules and regulations, which are subject to revision from time to time. While we have experience with these regulations, there can be no assurance that we will be able to fully comply with all applicable regulations.
Branded and Generic Pharmaceutical Products
All prescription pharmaceutical products distributed in the U.S., whether branded or generic, must be approved by the FDA. All applications for FDA approval must contain information relating to product formulation, raw material suppliers, stability, manufacturing processes, packaging, labeling, and quality control. Information to support the bioequivalence of generic drug products or the safety and effectiveness of new drug products for their intended use is also required to be submitted. There are generally two types of applications used for obtaining FDA approval of new products:
New Drug Application (“NDA”)—An NDA is filed when approval is sought to market a newly developed branded product and, in certain instances, for a new dosage form, a new delivery system, or a new indication for an approved drug.
Abbreviated New Drug Application (“ANDA”)—An ANDA is filed when approval is sought to market a generic equivalent of a drug approved under an NDA.
The ANDA development process is generally less time-consuming and less complex than the NDA development process. It typically does not require new preclinical and clinical studies, because it relies on the studies establishing safety and efficacy conducted for the branded drug approved through the NDA process. The ANDA process, however, typically requires one or more bioequivalence studies to show that the ANDA drug is bioequivalent to the previously approved reference listed drug (“RLD”).
7
The Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Act”) provides that generic drugs may enter the market after the approval of an ANDA, which requires (1) that bioequivalence to the branded product be demonstrated through clinical studies, and (2) either the expiration, invalidation or circumvention of any patents or the end of any other relevant market exclusivity periods related to the branded drug.
Accordingly, generic products generally provide a safe, effective, and cost-efficient alternative to users of branded products. Growth in the generic pharmaceutical industry has been driven by the increased market acceptance of generic drugs, as well as the number of branded drugs for which patent terms and/or other market exclusivities have expired.
Generic products are generally commercialized after the expiration of patent protection for the branded product and after the end of a period of non-patent market exclusivity. In addition to patent exclusivity, the holder of the NDA may be entitled to a period of non-patent market exclusivity, during which the FDA cannot approve an application for a generic product. Also, if the NDA is a new chemical entity (“NCE”), the FDA may not approve an ANDA for a generic product for up to five years following approval of the NDA for the NCE. If an NDA is not an NCE, but the holder of the NDA conducted clinical trials essential to approval of the NDA or a supplement thereto, the FDA may not approve a generic equivalent to the NDA for three years. Certain other periods of exclusivity may be available if the branded drug is indicated for treatment of a rare disease or is studied for pediatric indications.
In order to obtain FDA approval of NDAs and ANDAs, our manufacturing procedures and operations must conform to FDA requirements and guidelines, generally referred to as “cGMP.” The requirements for FDA approval encompass all aspects of the production process, including validation and recordkeeping, the standards around which are continuously changing and evolving. As a result, we must consistently monitor and comply with these changes.
Our facilities, procedures, operations, and testing of products are subject to periodic inspection by the FDA, the DEA, Health Canada, and other authorities. In addition, the FDA and Health Canada conduct pre-approval and post-approval reviews and plant inspections to determine whether our systems and processes are in compliance with cGMP and other FDA and Health Canada regulations. Our suppliers are subject to similar regulations and periodic inspections.
Controlled Substances
The DEA regulates certain drug products containing controlled substances, pursuant to the U.S. Controlled Substances Act (“CSA”). Certain of our products contain significant components that are classified as controlled substances. CSA and DEA regulations impose specific requirements on manufacturers and other entities that handle these substances including registration, recordkeeping, reporting, storage, security, and distribution. Recordkeeping requirements include accounting for the amount of product received, manufactured, stored, and distributed. Companies handling controlled substances also are required to maintain adequate security and to report suspicious orders, thefts, and significant losses. The DEA periodically inspects facilities for compliance with the CSA and its regulations. Failure to comply with current and future regulations of the DEA could lead to a variety of sanctions, including revocation or denial of renewal of DEA registrations, injunctions, or civil or criminal penalties.
In addition, we must submit a request to the DEA for a quota to purchase the amount of API needed to manufacture certain of our products deemed controlled substances. Without approved quotas from the DEA, we would not be able to purchase these ingredients from our suppliers. As a result, we are dependent upon the DEA to approve quotas large enough to support our continued manufacture of our controlled substances at commercial level. See “Risk Factors ― We are entirely dependent on periodic approval by the DEA for the supply of the API needed to manufacture our controlled substances. An inability to obtain such approvals would reduce or eliminate our revenues for our controlled substances, and could have a material adverse effect on our business, financial position, and operating results. In addition, we are subject to strict regulation by the DEA and are subject to sanctions if we are unable to comply with related regulatory requirements.”
Unapproved Products
Two of our products, EEMT and Opium Tincture, are marketed without approved NDAs or ANDAs. The FDA's policy with respect to the continued marketing of unapproved products appears in the FDA's September 2011
8
Compliance Policy Guide Sec. 440.100 titled “Marketed New Drugs without Approved NDAs or ANDAs.” Under this policy, the FDA has stated that it will follow a risk-based approach with regard to enforcement against marketing of unapproved products. The FDA evaluates whether to initiate enforcement action on a case-by-case basis, but gives higher priority to enforcement action against products in certain categories, such as those with potential safety risks or that lack evidence of effectiveness. We continue to believe that, so long as we comply with applicable manufacturing standards, the FDA will continue to operate on a risk-based approach and will not take action against us. However, we can offer no assurance that the FDA will continue to follow this approach or that it will not take a contrary position with any individual product or group of products. See “Risk Factors – Two of our products, which together comprised 7% of our total revenue in 2021, are marketed without approved NDAs or ANDAs and we can offer no assurances that the FDA will not require us to either seek approval for these products or withdraw them from the market. In either case, our business, financial position, and operating results could be materially adversely affected.”
Medicaid/Medicare
Medicaid and Medicare, both of which are U.S. federal health care programs administered by CMS, are major payors of pharmaceutical products, including those we produce.
Medicaid is administered by the states and jointly funded by the federal and state governments. Its focus is on low-income populations. State drug coverage policies under Medicaid may vary significantly state by state. The Patient Protection and Affordable Care Act (“PPACA”), as amended by the Health Care and Education and Reconciliation Act of 2010, together known as the Affordable Care Act (“ACA”), originally required states to expand their Medicaid programs to individuals with incomes up to 138% of the federal poverty level. Although the United States Supreme Court in 2011 made the Medicaid expansion optional, many states have expanded their Medicaid programs.
The ACA also made changes to Medicaid law that has negatively impacted our business. Pharmaceutical manufacturers that want their drug products covered by state Medicaid programs must enter into a rebate agreement with CMS and pay rebates to state Medicaid agencies on utilization of their drugs dispensed to Medicaid beneficiaries. The ACA raised the rebate percentages for both generic and branded pharmaceuticals effective January 1, 2010. The basic rebate is currently 13% of the average manufacturer price for sales of Medicaid-reimbursed products marketed under ANDAs. Sales of Medicaid-reimbursed products marketed under NDAs require manufacturers to rebate the greater of 23.1% of the average manufacturer price or the difference between the average manufacturer price and the “best price” (as defined in the Medicaid statute) during a specific period. In addition, there is an additional rebate if the average manufacturer price of the drug is rising faster than inflation. Federal and/or state governments may continue to enact measures aimed at reducing the cost of drugs to the Medicaid program.
Medicare is run by the federal government and is largely focused on the elderly and disabled. The Medicare Modernization Act of 2003 (“MMA”) created Medicare Part D to provide voluntary prescription drug coverage for Medicare beneficiaries. The MMA has increased the amount of reimbursement for pharmaceuticals, a trend that we believe will continue to benefit the generic pharmaceutical industry. The ACA made some changes to Part D to make it easier for Medicare beneficiaries to obtain drugs, such as reducing coinsurance amounts. The ACA also required pharmaceutical companies to provide discounts to Medicare Part D beneficiaries for the cost of branded prescription drugs. The ACA created a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% pursuant to the Bipartisan Budget Act of 2018, or BBA, effective as of 2019) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Under the Medicare Coverage Gap Discount Program, any pharmaceutical product marketed under an NDA, regardless of whether the product is marketed as a “generic,” is subject to the discount requirement. Our Candesartan Hydrochlorothiazide, Fenofibrate, Fluvoxamine, Hydrocortisone Enema, Lithium Carbonate ER, Mesalamine, Propranolol ER, Terbutaline, and Vancomycin products, while marketed as “generics,” are sold under approved NDAs and, therefore, are subject to the discount requirement.
Since its enactment, there have been judicial, administrative, executive and Congressional legislative challenges to certain aspects of the ACA. For example, the ACA is currently subject to a broad legal challenge in California vs. Azar before the U.S. Supreme Court. Additionally, in November 2020, the U.S. Supreme Court heard argument in Texas v.
9
Azar, which challenges the constitutionality of the ACA. Pending resolution of the litigation, all of the ACA, except for the individual mandate to buy health insurance remains in effect. Were the Supreme Court to invalidate the ACA, that could have far-reaching consequences of an uncertain nature for our industry. There are a number of additional bills pending in Congress and healthcare reform proposals at the state level that would affect drug pricing in the Medicare and Medicaid programs. This changing federal landscape has both positive and negative impacts on the U.S. healthcare industry with much remaining uncertain as to how various provisions of federal law, and potential modification or repeal of these laws, will ultimately affect the industry.
Most of our products are covered by Medicaid and Medicare. Our reporting and payment obligations under the Medicaid rebate program and other governmental purchasing and rebate programs are complex and may involve subjective decisions. Any determination that we have failed to comply with those obligations could subject us to penalties and sanctions, and we could be subject to federal or state false claims litigation.
There has also been recent heightened federal governmental scrutiny over the manner in which manufacturers set prices for their marketed products. For example, there have been several recent Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, a recent Presidential administration released a “Blueprint”, or plan, to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out of pocket costs of drug products paid by consumers.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing. We expect that additional state and federal health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Patents, Trademarks, and Licenses
We own the trademark names for most of our branded products, including Apexicon, Cortenema, Purified Cortrophin Gel, Cortrophin-Zinc, Inderal LA, Inderal XL, InnoPran XL, Lithobid, Reglan, Vancocin, and Veregen. We license the trademark names for Atacand, Atacand HCT, Arimidex, Casodex, Oxistat, and Pandel. With the exception of a license for patent technology for Inderal XL, InnoPran XL, and Veregen, we do not own or license any patents associated with these products. Further, patent protection and market exclusivity for these branded products have expired, with the exception of the Veregen product, which has three patents that expire in 2022, 2025, and 2026. Therefore, we consider the trademark names to be of material value and we act to protect these rights from infringement. However, our business is not dependent upon any single trademark. Trademark protection continues in some countries as long as used, and in other countries, as long as registered. Registration is for fixed terms and may be renewed indefinitely. We believe that sales of our branded products have benefited and will continue to benefit from the value of the product name.
We formerly received royalties from a license for patent rights initially owned by Cell Genesys, Inc., which merged with BioSante in 2009. The royalties were received as a result of sales and milestones related to the Yescarta® product. These royalties ceased after a final payment in 2021. See Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for further information.
10
Distribution Agreements
In addition to selling products under our own NDAs and ANDAs, we enter into marketing and distribution agreements with third parties in which we sell products under ANDAs or NDAs owned or licensed by these third parties. These products are sold under our own label.
Customers
Our customers purchase and distribute our products. Our products are sold by three major retail pharmacy chains: CVS, Rite Aid, and Walgreens. Our customers include five major national wholesalers: AmerisourceBergen, Cardinal Health, McKesson, Smith Drug Company, and Morris Dickson. In addition, our customers include national mail order houses, including CVS Caremark, Humana, and ExpressScripts, as well as group purchasing organizations.
In recent years, the wholesale distributor network for pharmaceutical products has been subject to increasing consolidation, which has increased the concentration of our wholesale customers. In addition, the number of retail market chains and, in particular, the number of independent drug stores and small chains, has decreased as retail consolidation has occurred, also increasing the concentration of our retail customers. As a result of this trend toward consolidation, a smaller number of companies each control a larger share of pharmaceutical distribution channels. For the year ended December 31, 2021, approximately 68% of our net revenues were attributable to three wholesalers: AmerisourceBergen Corporation, 29%, McKesson Corporation, 23%, and Cardinal Health, Inc., 16%. For the years ended December 31, 2020 and 2019, McKesson Corporation, Cardinal Health, Inc., and AmerisourceBergen Corporation, together accounted for approximately 74% and 80% of our net revenues, respectively. In addition, as noted below, our customers also distribute our products. The loss of any of these customers, including in their role as distributors, could have a material adverse effect on our business.
Due to a strategic partnership between Amerisource Bergen and Walgreens, Amerisource Bergen handles product distribution for Walgreens. Similarly, Cardinal Health and CVS established a partnership in which Cardinal performs some product distribution for CVS. McKesson also entered into a strategic alliance with both Wal-Mart and Rite Aid. As a result of these strategic partnerships between wholesalers and pharmacy chains, we have experienced, and expect to continue to experience, increases in net sales to the wholesalers, with corresponding decreases in net sales to the pharmacy chains.
In the rare disease business there is a limited distribution network and a select group of specialty pharmacies which can dispense product to appropriate patients. We are in the process of contracting with largest health insurance payers across the appropriate channels and classes of trade.
Consistent with industry practice, we maintain a return policy that allows customers to return product within a specified period prior to and subsequent to the expiration date. Generally, product may be returned for a period beginning six months prior to its expiration date to up to one year after its expiration date. See “Management’s Discussion and Analysis of Results of Operations and Financial Condition—Critical Accounting Estimates” for a discussion of our accruals for chargebacks, rebates, returns, and other allowances.
Sales, Marketing, and Distribution
We market, sell, and distribute our products in the United States. Our products are distributed through the following channels:
| ● | Wholesalers. We conduct business with five major wholesalers in the United States: AmerisourceBergen, Cardinal, McKesson, Smith Drug Company, and Morris Dickson. |
| ● | Retail Market Chains. We conduct business with three major retail chains in the United States: CVS, Rite Aid, and Walgreens. |
11
| ● | Distributors and Mail Order Pharmacies. We have contracts with several major distributors and mail order pharmacies in the United States, including Anda, CVS Caremark, Humana, and ExpressScripts. |
| ● | Group Purchasing Organizations. We have contracts with group purchasing organizations in the United States, such as ClarusONE, Walgreens Boots Alliance Development Group, Red Oak Sourcing, Econdisc, Optisource, Rx Sourcing Strategies, The Premier Group, Topco, The Buyer’s Consortium, Managed Health Care Associates Inc., Asembia, Premier Inc, and Kaiser Permanente. |
Competition
Certain of our products face limited competition due to complexities in formulation, active pharmaceutical ingredient sourcing, materials handling and manufacturing, and regulatory hurdles. Nevertheless, we compete with numerous other pharmaceutical companies, including large, global pharmaceutical manufacturers capable of addressing these complexities and hurdles with respect to products that we currently produce and products that are in our pipeline. In addition, our products are subject to competition from other generic products and non-prescription alternative therapies.
Our branded pharmaceutical products currently face competition from generic products and we expect them to continue to face competition from generic products in the future. In order to launch a generic product, a manufacturer must apply to the FDA for an ANDA showing that the generic product is therapeutically equivalent to the RLD. (See “Government Regulation.”)
The primary means of competition among generic drug manufacturers are pricing, contract terms, service levels, and reliability. To compete effectively, we seek to consistently produce high-quality, reliable, and effective products. We also establish active working relationships with each of our customers, continually gather important market information in order to respond successfully to requests for proposals, maintain sufficient inventories to assure high service levels, and work to reduce product costs by sourcing and qualifying alternative suppliers whenever possible.
Over the past several years, the pharmaceutical industry has experienced significant consolidation, particularly in distribution channels and among generic and brand drug companies.
The wholesale distributor network for pharmaceutical products has been subject to increasing consolidation, which has increased the concentration of our wholesale customers. In addition, the number of retail market chains and, in particular, the number of independent drug stores and small chains, has decreased as retail consolidation has occurred, also increasing the concentration of our retail customers. As a result of this trend toward consolidation, a smaller number of companies each control a larger share of pharmaceutical distribution channels, which results in pricing pressure on our business and can result in a shift in sales to our competitors.
In addition, consolidation among pharmaceutical companies has created opportunities by reducing the number of competitors. However, as competitors grow larger through consolidation, so do their resources. Larger competitors may be able to aggressively decrease prices in order to gain market share on certain products and may have resources that would allow them to market their products more effectively to potential customers.
Our sales can also be impacted by new studies that indicate that a competitor’s product has greater efficacy than one of our products. If competitors introduce new products with therapeutic or cost advantages, our products can be subject to progressive price reductions and/or decreased volume of sales.
Principal competitors for the pharmaceutical market in which we do business include Amneal Pharmaceuticals, Inc., Alvogen, Inc., Apotex Inc., Glenmark Pharmaceuticals Ltd, Hikma Pharmaceuticals plc, Method Pharmaceuticals, LLC, Viatris Inc., Par Pharmaceutical, Inc., Perrigo Company plc, Rising Pharmaceuticals, Inc., Sun Pharmaceutical Industries Ltd., and Teva Pharmaceuticals USA, Inc.
12
Product Liability
Product liability litigation represents an inherent risk to all firms in the pharmaceutical industry. We utilize traditional third-party insurance policies with regard to our product liability claims. Such insurance coverage at any given time reflects current market conditions, including cost and availability, when the policy is written.
Human Capital
As of January 2022, we have 601 employees, of which 457 are located in the United States, 123 are located in Canada, and 21 are located in India. We occasionally use a small number of part-time and consultant resources to meet our operational needs and are generally not impacted by significant turnover year-to-year. We are committed to creating a diverse and inclusive work environment within all levels of the business.
Attracting and retaining talented employees is critical to the success of our business, especially at our manufacturing operations in Baudette, Minnesota, which is located in a sparsely populated area of Northern Minnesota, with a population of less than 5,000. As a result, it can be challenging to find sufficiently qualified personnel in all functional areas. To address this, we support remote working arrangements for a number of employees in several functions throughout the business, including at the executive level. Additionally, our compensation plans are designed to be competitive within the pharmaceuticals industry as well as competitive with local employers for jobs of a cross-industry nature. Our approach provides ANI with the resources to recognize and reward employee performance, productivity, and quality commitment. Our total compensation program includes competitive base salaries, comprehensive benefits, and employee equity programs.
Our U.S., Canada, and India facilities are committed to the safety and health of our employees, patient-customers and the general public. It is critical within our mission to ensure we keep our employees and customers safe while accomplishing our business goals. We accomplish these initiatives through the following:
Health and Safety Management and Training
ANI has established a health and safety program with a focus on continuous improvement and employee engagement. ANI personnel are encouraged to take corrective actions where appropriate and to communicate concerns to management with a “see something, say something” approach. We recognize and reward personnel for contributing to the safety system within our working environment. The overall program continually evolves to reflect regulatory changes and compliance standard industry best practices. As part of onboarding new employees, we provide health and safety training and periodic training programs to maintain and improve employee awareness of safety issues. The goal of the safety training programs is to ensure that our staff are well informed on the subject matters and have the appropriate tools to make sound health and safety decisions in our day-to-day operations.
Environmental Stewardship
ANI is committed to minimizing waste and emissions, promoting reuse and recycling and conserving resources, where feasible, to reduce our environmental footprint on our environment.
COVID-19 Actions
Our U.S. and Canada facilities quickly responded to the COVID-19 pandemic by establishing a COVID-19 action plan to protect the health and safety of our employees as they performed their duties, as all of our facilities have remained open during the pandemic. Measures include social distancing requirements, increased and expanded sanitation for both employees and our property, staggered work schedules to minimize contact, flexible and work-from-home schedules, and employee illness and exposure protocols. These measures also seek to comply with all county, state, province, and/or city mandates as they relate to COVID-19. Please refer to Part I, Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of this Annual Report on Form 10-K for further discussion.
13
Available Information
We file annual, quarterly and current reports, proxy statements and other information required by the Securities Exchange Act of 1934, as amended (the “Exchange Act”), with the Securities and Exchange Commission (“SEC”). We make available free of charge on our website (www.anipharmaceuticals.com) our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements and any amendments to those filings as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. Also posted on our website in the “Investors – Corporate Governance” section are our Corporate Governance Guidelines, Code of Ethics and the charters for the Audit and Finance, Compensation, and Nominating and Corporate Governance Committees. Information on, or accessible through, our website is not a part of, and is not incorporated into, this report or any other SEC filing. Copies of our SEC filings or corporate governance materials are available without charge upon written request to Investor Relations, c/o ANI Pharmaceuticals, Inc., 210 Main Street West, Baudette, Minnesota, 56623.
14
Item 1A. Risk Factors
Risk Factor Summary
Investing in our common stock involves a high degree of risk. You should carefully consider all information in this Annual Report on Form 10-K prior to investing in our common stock. These risks are discussed more fully in the section titled “Risk Factors.” These risks and uncertainties include, but are not limited to, the following:
| ● | We may not achieve the anticipated benefits from our acquisition of Novitium Pharma LLC (“Novitium”) and we may face integration difficulties; |
| ● | The obligations and liabilities of Novitium, some of which may be unanticipated or unknown, may be greater than we have anticipated, which may diminish the value of Novitium to us; |
| ● | The uncertain impact that novel coronavirus (“COVID-19”) will have on our business and results of operations, including the emergence of variants of the virus; |
| ● | The continuing trend toward consolidation of customer groups that could result in declines in the sales volume and prices of our products, and increased fees charged by customers; |
| ● | Pharmaceutical product quality standards are steadily increasing on all products, and if we cannot meet these standards, we may be required to discontinue marketing and/or recall products from the market; |
| ● | Federal and state false claims litigation brought against us by private individuals and the government could result in civil and criminal penalties, damages, fines and other related actions; |
| ● | The use of legal, regulatory, and legislative strategies by competitors could result in increased costs to develop and market our products, delay new product introductions and reduce profit potential; |
| ● | Third-party payer actions may prevent us from effectively marketing our products or cause us to decrease pricing; |
| ● | Continuing studies of our products could produce results that could have a negative impact on our business; |
| ● | Healthcare reform legislation could have a material adverse effect on our business, financial position, and operating results; |
| ● | Barriers in achieving anticipated revenue growth and profitability could have a material adverse effect on our business, financial position, and operating results; |
| ● | Cortrophin Gel is our first rare disease pharmaceutical product and we recently announced commercial availability of this product. To the extent we are not able to achieve commercial success with this product, including gaining market share, our business, financial condition, and results of operations will be negatively impacted; |
| ● | The limited number of suppliers for our active pharmaceutical ingredients (“API”) could result in lengthy delays in production if we need to change suppliers; |
| ● | Several of the products we have acquired cannot be manufactured in our facilities and we must secure and maintain qualified and compliant contract manufacturers. Noncompliance by these contract manufacturers or our inability to find qualified contract manufacturers could result in us being unable to commercialize these products; Several of our products are manufactured and/or packaged by third parties, which we cannot control and could result in us being unable to market and distribute products; |
| ● | The FDA does not provide guidance on safety labeling for products that are marketed without approved NDAs or ANDAs, which could increase our potential liability with respect to failure-to-warn claims for these products; |
| ● | If the Drug Enforcement Administration (“DEA”) does not approve supply of the API we need to manufacture our controlled substances, we may be unable to manufacture controlled substances, which would eliminate our revenue on these products. |
15
| ● | Acquisitions and investments could disrupt our business and harm our financial position and operating results; |
| ● | Our Medicaid rebate accruals have increased and continue to increase due to our acquisitions and subsequent sales of branded products and authorized generics of branded products; |
| ● | Our accruals for the Medicare Coverage Gap Discount Program have increased due to growth and acquisitions; |
| ● | We face vigorous competition from other pharmaceutical manufacturers that threatens the commercial acceptance and pricing of our products; |
| ● | Our approved products, including Cortrophin Gel, may not achieve commercialization at levels of market acceptance that allow us to achieve profitability; |
| ● | We expect to spend a significant amount of resources on research and development efforts, and such efforts may not result in marketable products; |
| ● | Production at any or all of our four manufacturing facilities could be interrupted, which could cause us to fail to deliver product on a timely basis; |
| ● | We rely on third parties to assist with our clinical studies. If these parties do not perform or are non-compliant, it could negatively impact the clinical trial and potential of regulatory approval; Further, we may be required to audit or redo previously completed trials or recall already-approved commercial products; |
| ● | Inability to protect our intellectual property in the U.S. and foreign countries could negatively affect sales of our branded products; |
| ● | We have very limited staffing and are dependent upon key employees, the loss of whom could adversely affect our operations; |
| ● | We rely significantly on information technology and any failure, inadequacy, interruption, or security lapse of that technology could harm our ability to operate the business effectively; |
| ● | We are involved in and may become involved in legal proceedings from time to time, which may result in substantial losses, government enforcement actions, damage to our business and reputation, and place a strain on our internal resources; |
| ● | We are susceptible to product liability claims that may not be covered by insurance, which, if successful, could require us to pay substantial sums; |
| ● | Our policies regarding returns, allowances and chargebacks, and marketing programs adopted by wholesalers may reduce revenues in future fiscal periods; |
| ● | Making interest and principal payments under our Credit Agreement with Truist requires a significant amount of cash; |
| ● | Our Credit Facility contains restrictive and financial covenants and if are not in compliance with these covenants, our outstanding indebtedness under this facility could be accelerated and the lenders could terminate their commitments under the facility; |
| ● | Raising additional funds by issuing additional equity securities may cause dilution to our current stockholders. Raising additional funds by entering into additional credit or other borrowing facilities or issuing debt may subject us to covenants and other requirements that may restrict our operations; and |
| ● | Our international operations, including those resulting from our acquisition of Novitium and the global nature of its operations, will subject us to political and economic risks, increase our exposure to potential liability under anti-corruption, trade protection, tax, and other laws and regulations. |
16
The following are significant factors known to us that could materially harm our business, financial position, or operating results or could cause our actual results to differ materially from our anticipated results or other expectations, including those expressed in any forward-looking statement made in this report. The risks described are not the only risks facing us. Additional risks and uncertainties not currently known to us, or that we currently deem to be immaterial, also may adversely affect our business, financial position, and operating results. If any of these risks actually occur, our business, financial position, and operating results could suffer significantly. As a result, the market price of our common stock could decline and investors could lose all or part of their investment.
Risks Related to our Business
We may not achieve the anticipated benefits from our acquisition of Novitium and we may face integration difficulties, which could have a material adverse effect on our business, financial position, and operating results.
On November 19, 2021 (the “Closing Date”), the Company completed its previously announced acquisition (the “Acquisition”) of Novitium pursuant to the terms of the Agreement and Plan of Merger, dated as of March 8, 2021 (the “Merger Agreement”), by and among the Company, Novitium, Nile Merger Sub LLC, a Delaware limited liability company, and certain other parties, with Novitium becoming a wholly owned subsidiary of ANI.
We may not realize the potential benefits from the Acquisition that we or the market expects. Risks associated with the Acquisition include:
| • | failure to successfully integrate our businesses with the business of Novitium in the expected time frame which would adversely affect our financial condition and results of operation; |
| • | failure to effectively manage our expanded operations, which were materially increased by the Acquisition; |
| • | diversion of management’s attention, the disruption or interruption of, or the loss of momentum in, the businesses of ANI and Novitium or inconsistencies in standards, controls, procedures, and policies, any of which could adversely affect our ability to maintain relationships with customers, partners, and employees or our ability to achieve the anticipated benefits of the acquisition; |
| • | loss of key employees; and |
| • | failure to maintain relationships with third parties, including Novitium’s and our pre-existing customers, which relationships may be affected by customer preferences or public attitudes about the Acquisition. Any adverse changes in these relationships could adversely affect our business, financial condition, and results of operations. |
The obligations and liabilities of Novitium, some of which may be unanticipated or unknown, may be greater than we have anticipated, which may diminish the value of Novitium to us.
Novitium’s obligations and liabilities, some of which may not have been disclosed to us or may not be reflected or reserved for in Novitium’s historical financial statements, may be greater than we have anticipated. The obligations and liabilities of Novitium could have a material adverse effect on Novitium’s business or Novitium’s value to us or on our business, financial condition, or results of operations. Under the Merger Agreement relating to the Novitium acquisition, we have only limited indemnification with respect to obligations or liabilities of Novitium, whether known or unknown. In addition, even in cases where we are able to obtain indemnification, we may discover liabilities greater than the contractual limits or the financial resources of the indemnifying party. In the event that we are responsible for liabilities substantially in excess of any amounts recovered through rights to indemnification or alternative remedies that might be available to us, or any applicable insurance, we could suffer severe consequences that would substantially reduce our earnings and cash flows or otherwise materially and adversely affect our business, financial condition, or results of operations.
Our anticipated revenue growth and profitability, if achieved, is dependent upon our ability to develop, license or acquire, and commercialize new products on a timely basis in relation to our competitors’ product introductions, and to address all regulatory requirements applicable to the development and commercialization of new products. Our
17
failure to do so successfully could impair our growth strategy and plans and could have a material adverse effect on our business, financial position, and operating results.
Our future revenues and profitability are dependent upon our ability to successfully develop, license or acquire, and commercialize pharmaceutical products in a timely manner. Product development is inherently risky and time-consuming. Likewise, product licensing involves inherent risks, including uncertainties due to matters that may affect the achievement of milestones, as well as the possibility of contractual disagreements with regard to the supply of product meeting specifications and terms such as license scope or termination rights. The development and commercialization process also requires substantial time, effort, and financial resources. We may not be successful in commercializing products on a timely basis, if at all, which could adversely affect our business, financial position, and operating results.
The FDA must approve any new prescription product before it can be marketed in the U.S. The process of obtaining regulatory approval to manufacture and market branded and generic pharmaceutical products is rigorous, time consuming, costly, and largely unpredictable. We may be unable to obtain requisite approvals on a timely basis for branded or generic products that we may develop, license, or acquire. Moreover, if we obtain regulatory approval for a drug, we may be limited with respect to the indicated uses and delivery methods for which the drug may be marketed, which in turn could restrict the potential market for the drug. Also, for products pending approval, we may obtain raw materials or produce batches of inventory. In the event that regulatory approval is denied or delayed, we could be exposed to the risk of any such inventory becoming obsolete. The timing and cost of obtaining regulatory approvals could adversely affect our product introduction plans, business, financial position, and operating results.
The approval process for generic pharmaceutical products often results in the FDA granting simultaneous final approval to a number of generic pharmaceutical products at the time a patent claim for a corresponding branded product or other market exclusivity expires. This often forces a generic firm to face immediate competition when it introduces a generic product into the market. Additionally, further generic approvals often continue to be granted for a given product subsequent to the initial launch of the generic product. These circumstances generally result in significantly lower prices, as well as reduced margins, for generic products compared to branded products. New generic market entrants generally cause continued price and margin erosion over the generic product life cycle. As a result, we could be unable to grow or maintain market share with respect to our generic pharmaceutical products, which could have a material adverse effect on our ability to market that product profitably and on our business, financial position, and operating results.
Furthermore, if we are unable to address all regulatory requirements applicable to the development and commercialization of new products in a timely manner, our product introduction plans, business, financial position, and operating results could be materially adversely affected.
The FDA regulates and monitors all promotion and advertising of prescription drugs after approval. All promotion must be consistent with the conditions of approval and submitted to the agency. Failure to adhere to FDA promotional requirements can result in enforcement letters, warning letters, changes to existing promotional material, and corrective notices to healthcare professionals. Promotion of a prescription drug for uses not approved by the FDA can have serious consequences and result in lawsuits by private parties, state governments and the federal government, significant civil and criminal penalties, and compliance agreements that require a company to change current practices and prevent unlawful activity in the future.
Cortrophin Gel is our first rare disease pharmaceutical product, and we are developing a sales and marketing platform to commercialize this product. To the to the extent our efforts to commercialize this product are unsuccessful, our business, financial condition and results of operations will be negatively impacted.
On October 29, 2021, we received approval from the FDA for our Cortrophin Gel product for the treatment of certain chronic autoimmune disorders, including acute exacerbations of multiple sclerosis (“MS”) and rheumatoid arthritis (“RA”), in addition to excess urinary protein due to nephrotic syndrome. We have devoted significant time and money over the past five years to the development of this product since we acquired the rights to the product in 2016. We have invested and continue to invest significantly in the commercialization of this product in the U.S, including building out a sales force and developing a patient support program, with a full-scale launch in January 2022. The ability
18
for us to generate significant net product revenues from Cortrophin Gel will depend upon our ability to successfully sell the product and numerous other factors, including:
| ● | successfully establishing and maintaining effective sales, marketing, and distribution systems in jurisdictions in which Cortrophin Gel is approved for sale; |
| ● | successfully establishing and maintaining manufacturing capabilities and manufacturing adequate commercial quantities of Cortrophin Gel at acceptable cost and quality levels, including maintaining current good manufacturing practice (“cGMP”) and quality systems regulation standards required by various regulatory agencies; |
| ● | broad acceptance of Cortrophin Gel by physicians, patients and the healthcare community; |
| ● | the acceptance of pricing and placement of Cortrophin Gel on payers’ formularies and the associated tiers; |
| ● | effectively competing with the only other competitor that has an approved adrenocorticotropic hormone (“ACTH”) therapy product on the market, as well as other products that are in development or may be developed in the future as a treatment option; |
| ● | continued demonstration of safety and efficacy of Cortrophin Gel in comparison to competing products or treatment options; |
| ● | our ability to comply with ongoing regulatory obligations and continued regulatory review which may result in significant additional expense and may require labeling changes based on new safety information, post-market studies or clinical trials to evaluate safety risks related to the use of Cortrophin Gel; and |
| ● | obtaining, maintaining, enforcing, and defending intellectual property rights and claims. |
If we do not achieve one or more of these factors, we could experience an inability to successfully commercialize Cortrophin Gel, which would negatively impact our business, financial condition and results of operations. In addition, sales of Cortrophin Gel could be negatively affected by discovery of previously unknown problems with the product, such as adverse events of unanticipated severity or frequency, problems with the facilities where the product is manufactured, or imposition of restrictions on Cortrophin Gel, including requiring withdrawal of the product from the market, by a regulatory agency if it disagrees with the promotion, marketing, or labeling of the product.
We are developing our marketing and sales organization to support Cortrophin Gel and have no experience in marketing prescription rare disease drug products. If we are unable to successfully establish marketing and sales capabilities for Cortrophin Gel, our business will suffer.
We have only recently established rare disease sales, marketing or distribution capabilities and have no institutional experience in marketing rare disease products. We intend to continue to develop an in-house marketing organization and sales force, which will require significant expenditures, management resources and time. We will have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train and retain marketing and sales personnel.
We depend on a limited number of suppliers for API. Generally, only a single source of API is qualified for use in each product due to the costs and time required to validate a second source of supply. We may experience lengthy delays if we need to change an API supplier, which could have a material impact on business and results of operations.
Our ability to manufacture and distribute products is dependent, in part, upon ingredients and components supplied by others, including entities based outside the U.S. During the year ended December 31, 2021, no single vendor represented at least 10% of inventory purchases. We purchased approximately 10% and 13% of our inventory from one supplier during the years ended December 31, 2020 and 2019, respectively. Any disruption in the supply of these ingredients or components or any problems in their quality could materially affect our ability to manufacture and distribute our products and could result in legal liabilities that could materially affect our ability to realize profits or otherwise harm our business, financial, and operating results. Virtually all of our generic contracts for the supply of pharmaceutical products to customers contain "failure to supply" clauses. Under these clauses, if we are unable to supply the requested quantity of product within a certain period after receipt of a customer’s purchase order, the customer is entitled to procure a substitute product elsewhere and we must reimburse the customer for the difference between our contract price and the price the customer was forced to pay to procure the substitute product. Therefore, our ability to source sufficient quantities of API for manufacturing is critical. We source the raw materials for our products from both
19
domestic and international suppliers. Generally, we qualify only a single source of API for use in each product due to the cost and time required to validate and qualify a second source of supply. Any change in one of our API suppliers must usually be approved through a Prior Approval Supplement (“PAS”) by the FDA. The process of obtaining an approval of such a PAS can require between 4 and 18 months. While we also generally qualify a single source for non-API raw materials, the process required to qualify an alternative source of a non-API raw material is typically much less rigorous. If we were to change the supplier of a raw material for a product, the cost for the material could be greater than the amount we paid with the previous supplier. Changes in suppliers are rare but could occur as a result of a supplier’s business failing, an issue arising from an FDA inspection, or failure to maintain our required standards of quality. As a result, we carefully select suppliers, based on various factors including quality, reliability of supply, and long-term financial stability. Certain of the APIs for our drug products, including those that are marketed without approved NDAs or ANDAs, are sourced from international suppliers. From time to time, we have experienced temporary disruptions in the supply of certain of such imported API due to FDA inspections. In addition, the COVID-19 pandemic and associated workforce factors has disrupted certain supply chains and generally led to longer lead times for the procurement of goods that are essential to the manufacture of our products.
Several of the products we have acquired cannot be manufactured in our facilities and are manufactured and/or packaged by third parties, which we cannot control. If we are unable to secure or maintain qualified contract manufacturers for those products or if a contract manufacturer fails to comply with federal, state, and local laws and regulations, our business, financial position, and operating results could be materially, adversely affected.
We have acquired, and may continue to acquire, a variety of products that we seek to commercialize. Some of these products, including injectables, softgel capsules, and Purified Cortrophin Gel, are products that we cannot currently manufacture in our facilities. As a result, we may seek partners to contract manufacture the products on our behalf, and we rely on third parties to manufacture and/or package many of our products. Like our company, these firms must comply with cGMPs and other federal, state, and local laws and regulations regarding pharmaceutical manufacturing. Noncompliance by those firms may result in warning letters, fines, product recalls, and partial or total suspension of production and distribution. If we are unable to find qualified contract manufacturers or if a contract manufacturer fails to comply with federal, state, and local laws and regulations, we may be unable to commercialize these products, which could have a material adverse effect on our business, financial position, and operating results, including an impairment of the acquired product.
We expect our reliance on third party manufacturers to continue to increase in the future as we receive approvals for new products to be manufactured through our collaborative arrangements, and as we seek additional growth opportunities outside of the capabilities of our current manufacturing facilities. If we are unable to secure third-party manufacturers for these products on commercially acceptable terms, we may not be able to market and distribute such products at a profit. Any delays or difficulties with third-party manufacturers could adversely affect the marketing and distribution of these products, or future products, which could have a material adverse effect on our business, financial position, and operating results.
Our branded products may become subject to increased generic competition.
Many of our branded products have not been patent-protected for several years and no longer have market exclusivity. As a result, they face competition from lower priced generic products which may reduce and limit the sales of our mature brand products. Additionally, increased focus by the FDA on approval of generic products may accelerate this trend. If generic products are substituted for these branded products, our revenue from these products will decrease, which could have an adverse effect on our business, financial position, and operating results.
Future acquisitions and investments could disrupt our business and harm our financial position and operating results.
Our growth will depend, in part, on our continued ability to develop, commercialize, and expand our products, including in response to changing regulatory and competitive pressures. In some circumstances, we have and may continue to grow our business through the acquisition of complementary businesses and technologies rather than through internal development. The identification of suitable acquisition candidates or products can be difficult, time-consuming,
20
and costly, and we may not be able to successfully complete or successfully execute strategies for identified acquisitions. The risks faced in connection with acquisitions include:
| ● | diversion of management time and focus from operating our business to addressing acquisition and/or product integration challenges; |
| ● | coordination of research and development and sales and marketing functions; |
| ● | retention of key employees from the acquired company; |
| ● | integration of the acquired company’s accounting information, management, human resources, and other administrative systems; |
| ● | the need to implement or improve controls, procedures, and policies at a business that prior to the acquisition may have lacked effective controls, procedures and policies; |
| ● | difficulties relating to integrating the acquired business; |
| ● | liability for activities of the acquired company and/or products before the acquisition, including patent infringement claims, violations of laws, commercial disputes, tax liabilities and other known and unknown liabilities; |
| ● | unanticipated write-offs or charges; and |
| ● | litigation or other claims in connection with the acquired company or product, including claims from product users, former stockholders, or other third parties. |
In any acquisition that we may undertake, our failure to address these risks or other problems encountered in connection with any acquisitions and investments could cause us to fail to realize the anticipated benefits of these acquisitions or investments, cause us to incur unanticipated liabilities, and harm our business generally.
Our Medicaid rebate accruals have increased and continue to increase due to our acquisitions and subsequent sales of branded products and authorized generics of branded products, and the estimates on which our accruals are based are subject to change. Any such change could have a material adverse effect on our business, financial position, and operating results.
Our Medicaid rebate accruals have increased significantly due to our acquisitions and subsequent sales of branded products and authorized generics of branded products. We accrue for these rebates at the time of sale based on our estimates of the amount of our product that will be prescribed to Medicaid beneficiaries. The resulting accruals are significant, and as Medicaid utilization trends change, we may need to change our estimates accordingly. We cannot guarantee that actual results will not differ from our estimates. In addition, the Patient Protection and Affordable Care Act (“PPACA”) included a significant expansion of state Medicaid programs. As more individuals become eligible for coverage under these programs, Medicaid utilization of our products could increase, resulting in a corresponding increase in our rebate payments. Increases in Medicaid rebate payments could decrease our revenues from product sales, which in turn could adversely affect our business, financial position, and operating results.
Our accruals for the Medicare Coverage Gap Discount Program have increased due to growth and acquisitions. Any such change could have a material adverse effect on our business, financial position, and operating results.
Our accruals for the rebates under the Medicare Coverage Gap Discount Program have increased due to growth and acquisitions. We accrue for these rebates at the time of sale based on our estimates of the amount of product that will be prescribed to patients in the Medicare Coverage Gap Discount program, which is primarily for the benefit of persons aged 65 years and over. As we acquire and launch additional products, many of which, are often used by patients in the 65 and older age range, our estimates of these rebates have grown. Increases in Medicare Coverage Gap Discount rebates could decrease our revenues from product sales, which in turn could adversely affect our business, financial position, and operating results.
21
We have entered into distribution agreements under which we market products under ANDAs and NDAs owned by third parties. Any changes to these agreements could have a material adverse effect on our business, financial position, and operating results.
We have entered into several distribution agreements to market and distribute products under our own label that are sold under ANDAs and NDAs owned by third parties, over which we have no control. Generally, the responsibility for maintaining the ANDAs and NDAs lies with these third parties. If any regulatory issues were to arise with the underlying ANDA or NDA for one of these products, we could be required to discontinue sales of the product, which could have an adverse effect on our business, financial position, and operating results.
We face vigorous competition from other pharmaceutical manufacturers that may adversely impact commercial acceptance and pricing of our products. If we are unable to successfully compete, such competition could have a material adverse effect on our business, financial position, and operating results.
The generic pharmaceutical industry is highly competitive. We face intense competition from U.S. and foreign manufacturers, many of whom are significantly larger than us and operate in lower cost geographies. Our competitors may be able to develop products and processes competitive with or superior to ours for many reasons, including but not limited to the possibility that they may have:
Any of our significant competitors, due to one or more of these and other factors, could have a material adverse effect on our business, financial position, and operating results.
Our approved products may not achieve commercialization at levels of market acceptance that allow us to achieve profitability, which could have a material adverse effect on our business, financial position, and operating results.
We seek to develop, license, or acquire products that we can commercialize at levels of market acceptance that would allow us to recoup our costs, grow market share, and achieve profitability. Even if we are able to obtain regulatory approvals for our pharmaceutical products, if we fail to predict accurately demand for such products, our business, financial position, and operating results could be adversely affected. Levels of market acceptance for our products could be impacted by several factors, including but not limited to:
Some of these factors are outside of our control and, if any arise, our profitability, business, financial position, and operating results could be materially adversely affected.
We have entered into several collaborative arrangements that may not result in marketable products.
We have entered into several collaborative arrangements to develop generic products for us to market in the U.S. We can offer no assurances that these arrangements will result in additional approved products, or that we will be able to
22
market the products at a profit. In addition, any expenses related to clinical trials, or additional studies required by the FDA, that we may incur in connection with these collaborative arrangements may negatively affect our business, financial position, and operating results. Specifically:
Any of these events could have a material adverse effect on our business, financial position, and operating results.
We expect to spend a significant amount of resources on research and development efforts, and such efforts may not result in marketable products. Failure to successfully introduce products into the market could have a material adverse effect on our business, financial position, and operating results.
We conduct research and development primarily to enable us to manufacture and market approved products in accordance with applicable regulations. Research and development is expensive and time-consuming. As we seek to develop new products, or re-commercialize products that were previously approved, our research expenses will increase, potentially significantly, and we cannot be certain that we will recover our investment in a product, even if that product is commercialized. If we spend significant resources on research and development efforts and are not able to introduce new products, our business, financial position, and operating results may be materially adversely affected.
We own four manufacturing facilities that produce the majority of our products. Production at any or all of these facilities could be interrupted, which could cause us to fail to deliver sufficient product to customers on a timely basis and have a material adverse effect on our business, financial position, and operating results.
Our manufacturing operations are based in four facilities. While these facilities are sufficient for our current needs, the facilities are highly specialized and any damage to or need for replacement of all or any significant function of our facilities could be very costly and time-consuming and could impair or prohibit production and shipping. A significant disruption at any of the facilities, even on a short-term basis, whether due to a labor strike, adverse quality or compliance observation, vandalism, natural disaster, fire, storm or other environmental damage, or other events could impair our ability to produce and ship products on a timely basis and, among other consequences, could subject us to “failure to supply” claims from our customers, as discussed below. Although we believe we carry commercially reasonable business interruption and liability insurance, we might suffer losses because of business interruptions that exceed the coverage available under our insurance policies or for which we do not have coverage. Any of these events could have a material adverse effect on our business, financial position, and operating results.
Virtually all our contracts for the supply of generic products to our customers contain "failure to supply" clauses which require us to reimburse the customer for the difference between our contract price and the price the customer was forced to pay to procure the substitute product in the event we failed to deliver the requested quantity within a specified period of time. This difference can be substantial because of the much higher spot price at which the customer must cover its requirements and can be far in excess of the revenue that we would otherwise have received on the sale of our own product. Therefore, our ability to produce and ship a sufficient quantity of product on a consistent basis is critical. Failure to deliver products could have a material adverse effect on our business, financial position, and operating results.
We rely on third parties to assist with our clinical studies. If these third parties do not perform as required or expected, or if they are not in compliance with FDA rules and regulations, our clinical studies may be extended, delayed or terminated, or may need to be repeated, and we may not be able to obtain regulatory approval for or commercialize the products being tested in such studies. Further, we may be required to audit or redo previously completed trials or recall already-approved commercial products.
We rely on third parties, such as medical institutions, clinical investigators, and contract laboratories, to assist with our clinical studies. We are responsible for confirming that our studies are conducted in accordance with applicable
23
regulations and that each of our clinical studies is conducted in accordance with our general investigational plan and protocol. The FDA requires us to comply with regulations and standards, commonly referred to as good clinical practices for conducting, monitoring, recording, and reporting the results of clinical studies, to assure that data and reported results are accurate and that the clinical study participants are adequately protected. Our reliance on these third parties does not relieve us of these responsibilities. If the third parties assisting us with our clinical studies do not perform their contractual duties or obligations, do not meet expected deadlines, fail to comply with the FDA’s good clinical practice regulations, do not adhere to our protocols or otherwise fail to generate reliable clinical data, we may need to enter into new arrangements with alternative third parties and our clinical studies may be extended, delayed or terminated or may need to be repeated, and we may not be able to obtain regulatory approval for or commercialize the products being tested in such studies. For our already-approved commercial products, we may be required to audit or redo previously completed trials or recall our products from the market, which could have a material adverse effect on our business, financial position, and operating results.
With the exception of a license of patent technology for Veregen we do not own or license any material patents associated with our products, and our ability to protect and control unpatented trade secrets, know-how, and other technological innovation is limited.
Generally, the branded pharmaceutical business relies upon patent protection to ensure market exclusivity for the life of the patent. Except for a license for patent technology for Veregen we do not own or license any material patents associated with our products and therefore do not enjoy the same level of intellectual property protection with respect to such products as would a pharmaceutical manufacturer that markets a patented product. We have limited ability to protect and control trade secrets, know-how, and other technological innovation, all of which are unpatented. Others independently may develop similar or better proprietary information and techniques and disclose them publicly. In addition, others may gain access to our trade secrets, and we may not be able to protect our rights to our unpatented trade secrets. In addition, confidentiality agreements and other measures may not provide protection for our trade secrets in the event of unauthorized use or disclosure of such information. Failure to protect and control such trade secrets, know-how and innovation could harm the value of our trade secrets, know-how and other technological innovation, which could have a material adverse effect on our business, financial position, and operating results.
Inability to protect our intellectual property in the U.S. and foreign countries could negatively affect sales of our branded products.
We own the trademark names for most of our branded products, including, Apexicon, Cortenema, Purified Cortrophin Gel, Cortrophin-Zinc, Inderal LA, Inderal XL, InnoPran XL, Lithobid, Reglan, Vancocin, and Veregen. We license the trademark names for Atacand, Atacand HCT, Arimidex, Casodex, Oxistat, and Pandel. While we will seek to protect those trademarks through timely renewal in applicable jurisdictions, we may not be able to renew our trademarks in a timely manner or to prevent third parties from using our trademarks, which could have a material adverse effect on our business, financial position, and operating results.
We have very limited staffing and are dependent upon key employees, the loss of whom could adversely affect our operations. Competition for talent is intense, especially in northern Minnesota, where the population is small. If we cannot attract and retain qualified personnel, the growth and success of our business could be adversely affected.
Our success is dependent upon the efforts of a relatively small management team and staff. We have employment arrangements in place with our executive and other officers, but none of these executive and other officers are bound legally to remain employed with ANI for any specific term. We do not have key person life insurance policies covering our executive and other officers or any of our other employees. If key individuals were to leave ANI, our business could be affected adversely if suitable replacement personnel are not recruited quickly. The population in northern Minnesota, where two of our four manufacturing facilities are located, is small, and as a result, there is a limited number of qualified personnel available in all functional areas, which could make it difficult to retain and attract the qualified personnel necessary for the development and growth of our business. If we were unable to attract and retain qualified personnel, our business, financial position, and operating results could be materially adversely affected.
24
We rely significantly on information technology and any failure, inadequacy, interruption, or security lapse of that technology, including any cybersecurity incidents, could harm our ability to operate the business effectively.
We rely significantly on our information technology and manufacturing infrastructure to effectively manage and maintain inventory and financial reports, manufacture and ship products, and invoice customers in a timely manner. While we have invested in the protection of data and information technology, any failure, accidents, inadequacy, or interruption of that infrastructure or security lapse of that technology, including cybersecurity incidents, could harm our ability to operate our business effectively. Our ability to manage and maintain inventory and financial reports, manufacture and ship products, and invoice customers timely depends significantly on our general ledger, our contracted electronic data interface system, and other information systems. Cybersecurity attacks in particular are evolving and include, but are not limited to, malicious software, attempts to gain unauthorized access to data and other electronic security breaches that could lead to disruptions in systems, misappropriation of confidential or otherwise protected information and corruption of data. Cybersecurity incidents resulting in the failure of our information systems to operate effectively or to integrate with other systems, or a breach in security or other unauthorized access of these systems, may affect our ability to manage and maintain inventory and financial reports, and result in delays in product fulfillment and reduced efficiency of operations. A breach in security, unauthorized access resulting in misappropriation, theft, or sabotage with respect to proprietary and confidential information, including research or clinical data, could require significant capital investments to remediate any such failure, problem or breach, all of which could adversely affect our business, financial position, and operating results.
We are currently involved in and may from time to time become involved in legal proceedings, some of which may result in substantial losses, government enforcement actions, damage to our business and reputation, and place a strain on our internal resources.
We are currently involved in and in the future may become involved in legal proceedings in the ordinary course of our business, as a party or non-party witness, with both private parties and certain government agencies. We may incur substantial time and expenses participating in these types of lawsuits and investigations, which could also divert management’s attention from ongoing business concerns and normal operations. In addition, these matters and any other substantial litigation may result in verdicts against us or government enforcement actions, which may include significant monetary awards, and preventing the manufacture, marketing and sale of our products. Any dispute resolved unfavorably, could have a material adverse effect on our business, financial position, and operating results. For a description of legal proceedings which are currently pending, see Note 12. Commitments and Contingencies, in the notes to the consolidated financial statements in Part II, Item 8 of this Annual Report on Form 10-K.
We are susceptible to product liability claims that may not be covered by insurance, which, if successful, could require us to pay substantial sums.
Like all pharmaceutical companies, we face of the risk of loss resulting from, and the adverse publicity associated with, product liability lawsuits, whether or not such claims are valid. We likely cannot avoid such claims. Unanticipated side effects or unfavorable publicity concerning any of our products or product candidates would likely have an adverse effect on our ability to achieve acceptance by prescribing physicians, managed care providers, pharmacies and other retailers, customers, patients and clinical trial participants. Even unsuccessful product liability claims could require us to spend money on litigation, divert management’s time, damage our reputation and impair the marketability of our products. In addition, although we believe that we have adequate product liability insurance coverage, we cannot be certain that our insurance will, in fact, be sufficient to cover such claims or that we will be able to obtain or maintain adequate insurance coverage in the future at acceptable prices. A successful product liability claim that is excluded from coverage or exceeds our policy limits could require us to pay substantial sums. Additionally, insurance coverage for product liability may become prohibitively expensive in the future or may not be available at all, and as a result, we may not be able to maintain adequate product liability insurance coverage to mitigate the risk of large claims, or we may be required to maintain a larger self-insured retention that we would otherwise choose.
25
Currency fluctuations and changes in exchange rates could have a material adverse effect on our business, financial position, and operating results.
A portion of our transactions are denominated in a foreign currency, the Canadian dollar and the Indian rupee. Because we engage in certain transactions in a foreign currency, we are subject to the effects of exchange rate fluctuations. If the U.S. dollar depreciates against the Canadian dollar and the Indian rupee, the expenses we recognize from Canadian-denominated and Indian-denominated transactions made by our Canadian and Indian subsidiaries could be translated at an unfavorable rate, leading to foreign exchange losses. Foreign exchange gains or losses as a result of exchange rate fluctuations in any given period could harm our operating results and negatively impact our financial position and results of operations.
Risks Related to our Industry
The COVID-19 pandemic is ongoing and its impact on the global economy and our operations remains uncertain. A continuation of the pandemic could have a material adverse impact on our business, results of operations and financial condition and on the market price of our common stock.
On March 12, 2020, the World Health Organization declared COVID-19 to be a pandemic. In an effort to contain and mitigate the spread of COVID-19, many countries, including the United States and Canada, imposed unprecedented restrictions on travel, and there were business closures and a substantial reduction in economic activity in countries that have had significant outbreaks of COVID-19. While restrictions and impacts eased in 2021, significant uncertainty remains as to the continued potential impact of the COVID-19 pandemic on our operations and on the global economy as a whole.
Demand for the products we sell was negatively impacted by COVID-19 during the years ended December 31, 2021 and 2020, as fewer patients visited physicians for conditions treated by our products, fewer elective surgeries occurred and visits to pharmacies declined due to government orders and closures of or restrictions placed on visits to medical offices and facilities. Additionally, we have experienced disruptions to our supply chain, including increased lead times on the procurement of materials. While most government orders, closures and restrictions have now lapsed, this situation could continue or worsen depending on the duration and severity of the COVID-19 pandemic, the level of success in implementing mitigation measures, such as vaccines, the continued emergence of new variants of COVID-19, the length of time it takes for normal economic and operating conditions to resume, the impact of the pandemic on inflation, additional governmental actions that may be taken, and numerous other uncertainties.
It is currently not possible to predict how long the pandemic will continue, whether new government restrictions will be reinstituted, the effectiveness of mitigation efforts such as vaccines, the emergence of new variants of the virus, and the related impact on economic activity, including inflation. A disruption in the financial markets and volatility, as seen in 2020 and 2021, could have an adverse effect on our ability to access capital, our pharmaceutical supply chain, our business, results of operations and financial condition, and the market price of our common stock.
The continuing trend toward consolidation of customer groups could result in declines in the sales volume and prices of our products, and increased fees charged by customers, each of which could have a material adverse effect on our business, financial position, and operating results.
Consolidation and the formation of strategic partnerships among and between wholesale distributors, chain drug stores, and group purchasing organizations has resulted in a smaller number of companies, each controlling a larger share of pharmaceutical distribution channels. For example, our net revenues are concentrated among three customers representing 29%, 23%, and 16% of net revenues, respectively, during the year ended December 31, 2021. As of December 31, 2021, accounts receivable from these three customers was approximately 92% of our accounts receivable, net. Drug wholesalers and retail pharmacy chains, which represent an essential part of the distribution chain for generic pharmaceutical products, have undergone, and are continuing to undergo, significant consolidation. This consolidation may result in declines in our sales volumes if a customer is consolidated into another company that purchases products from a competitor. In addition, the consolidation of drug wholesalers and retail pharmacy chains could result in these groups gaining additional purchasing leverage and consequently increasing the product pricing pressures facing our business and enabling those groups to charge us increased fees. Additionally, the emergence of large buying groups
26
representing independent retail pharmacies and the prevalence and influence of managed care organizations and similar institutions potentially enable those groups to extract price discounts on our products. The result of these developments or the loss of our relationship with one or more of these wholesalers, may have a material adverse effect on our business, financial position, and operating results.
Our reporting and payment obligations under the Medicaid rebate program and other governmental purchasing and rebate programs are complex and may involve subjective decisions. Any determination that we have failed to comply with those obligations could subject us to penalties and sanctions, which could adversely affect our business, financial position, and operating results.
The regulations regarding reporting and payment obligations with respect to Medicaid rebates and other governmental programs are complex. Because our processes for these calculations and the judgments involved in making these calculations involve subjective decisions and complex methodologies, these calculations are subject to the risk of errors. Our calculations and methodologies are subject to review and challenge by governmental agencies, and it is possible that such reviews could result in changes. Any determination by governmental agencies that we have failed to comply with our reporting and payment obligations could subject us to penalties and sanctions, which could have a material adverse effect on our business, financial position, and operating results.
Two of our products, which together comprised 7% of our total revenue in 2021, are marketed without approved NDAs or Abbreviated New Drug Applications (“ANDAs”) and we can offer no assurances that the U.S. Food and Drug Administration (“FDA”) will not require us to either seek approval for these products or withdraw them from the market. In either case, our business, financial position, and operating results could be materially adversely affected.
Two of our products, Esterified Estrogen with Methyltestosterone (“EEMT”) and Opium Tincture, are marketed without approved NDAs or ANDAs.
The FDA's policy with respect to the continued marketing of unapproved products appears in the FDA's September 2011 Compliance Policy Guide Sec. 440.100 titled “Marketed New Drugs without Approved NDAs or ANDAs.” Under this policy, the FDA has stated that it will follow a risk-based approach with regard to enforcement against marketing of unapproved products. The FDA evaluates whether to initiate enforcement action on a case-by-case basis, but gives higher priority to enforcement action against products in certain categories, such as those with potential safety risks or that lack evidence of effectiveness.
We continue to believe that, so long as we comply with applicable manufacturing standards, the FDA will continue to operate on a risk-based approach and will not take action against us. However, we can offer no assurance that the FDA will continue to follow this approach or that it will not take a contrary position with any individual product or group of products.
Additionally, our EEMT products are related to an outstanding Notice of Opportunity for Hearing on estrogen-androgen products. The hearing relates to the FDA's intent to reclassify certain estrogen-androgen combination drugs as lacking substantial evidence of their effectiveness for the treatment of moderate to severe vasomotor symptoms associated with the menopause in those patients not improved by estrogen alone.
If the FDA were to move away from the risk-based approach to enforcement against marketing of unapproved products, we may be required to seek FDA approval for these products or withdraw such products from the market. If we decide to withdraw the products from the market, our net revenues for generic pharmaceutical products would decline materially, and if we decide to seek FDA approval, we would face increased expenses and might need to suspend sales of the products until such approval was obtained, and there are no assurances that we would receive such approval.
27
Imported API are subject to inspection by the FDA and the FDA can refuse to permit the importation of API for use in products that are marketed without approved NDAs or ANDAs. We are dependent on imported API to make certain of our products. If the FDA detained or refused to allow the importation of such API, our revenues from certain of our products would be reduced or eliminated and our business, financial position, and operating results could be materially adversely affected.
We source some of the API for our products, including those that are marketed without approved NDAs or ANDAs, from international suppliers. From time to time, due to FDA inspections, we have experienced temporary disruptions in the supply of imported API. Any prolonged disruption in the supply of imported API could materially affect our ability to manufacture and distribute our products, reduce or eliminate our revenues, and have a material adverse effect on our business, financial position, and operating results. In addition, as regulatory fees and compliance oversight of API manufacturers increase, this could result in certain companies discontinuing their supply of API to us, which would materially affect our ability to manufacture our products.
The FDA does not provide guidance on safety labeling for products that are marketed without approved NDAs or ANDAs. As a result, we are dependent on our internal post-approval drug safety surveillance program to identify necessary safety-related changes to the labels for EEMT and Opium Tincture.
Pharmaceutical product labels contain important safety information including Black Box warnings, contraindications, dosing and administration, adverse reactions, drug interactions, use in specific populations such as pregnant women, pediatric, and geriatric patients, and other warnings and precautions. Pharmaceutical manufacturers may change product labels when post-approval drug safety surveillance programs identify previously unknown side-effects, drug interactions, and other risks. Manufacturers may also change product labels after conducting post-approval clinical studies and may receive or seek guidance from the FDA regarding updating safety labeling information. However, the FDA does not provide guidance on labeling for products that are marketed without approved NDAs or ANDAs. As a result, we are dependent on our internal post-approval drug safety surveillance program to identify necessary safety-related changes to the labels for EEMT and Opium Tincture. Additionally, because the FDA does not review and approve labeling for the products without approved NDAs or ANDAs, it would be difficult to make a claim for preemption due to the FDA’s approval of the labeling and this could increase our potential liability with respect to failure-to-warn claims for these products. Such claims, even if successfully defended, could have an adverse impact on our business, financial position, and operating results.
We are entirely dependent on periodic approval by the DEA for the supply of the API needed to manufacture our controlled substances. An inability to obtain such approvals would reduce or eliminate our revenues for our controlled substances, and could have a material adverse effect on our business, financial position, and operating results. In addition, we are subject to strict regulation by the DEA and are subject to sanctions if we are unable to comply with related regulatory requirements.
The DEA regulates products containing controlled substances, such as opiates, pursuant to the U.S. Controlled Substances Act (“CSA”). The CSA and DEA regulations impose specific requirements on manufacturers and other entities that handle these substances including registration, recordkeeping, reporting, storage, security, and distribution. Recordkeeping requirements include accounting for the amount of product received, manufactured, stored, and distributed. Companies handling controlled substances also are required to maintain adequate security and to report suspicious orders, thefts and significant losses. The DEA periodically inspects facilities for compliance with the CSA and its regulations. Failure to comply with current and future regulations of the DEA could lead to a variety of sanctions, including revocation or denial of renewal of DEA registrations, injunctions, or civil or criminal penalties.
In addition, each year, we must submit a request to the DEA for a procurement quota in order to purchase the amount of API needed to manufacture our Schedule II controlled substances. Without approved procurement quotas from the DEA, we would not be able to purchase these ingredients from our suppliers. As a result, we are entirely dependent upon the DEA to approve, on an annual basis, a quota of API that is sufficiently large to support our plans for the continued manufacture of our controlled substances at commercial levels. In 2017, the DEA announced that the administration would decrease the total quotas approved for Schedule II opioid painkillers. In 2018, the DEA decreased quotas approved for Schedule II opioid painkillers. The DEA continues to closely monitor quotas of certain opioids and as a result there may be a reduction from what was requested; however, firms may file an application for a quota
28
adjustment at any time during the calendar year. If the DEA does not approve our requested procurement quotas, we may be unable to obtain sufficient API to manufacture these products at levels required by our customers, which could have an adverse impact on our business, financial position, and operating results.
Pharmaceutical product quality standards are steadily increasing and all products, including those already approved, may need to meet current standards or enhanced standards in the future. If our products are not able to meet these standards, we may be required to discontinue marketing and/or recall such products from the market.
Steadily increasing quality standards are applicable to pharmaceutical products still under development and those already approved and on the market. These standards result from product quality initiatives implemented by the FDA, such as criteria for residual solvents, periodic guidance from the FDA regarding testing for impurities, such as nitrosamines, in our products, and updated U.S. Pharmacopeial Convention (“USP”) Reference Standards. The USP is a scientific nonprofit organization that sets standards for the identity, strength, quality, and purity of medicines, food ingredients, and dietary supplements manufactured, distributed, and consumed worldwide. Pharmaceutical products approved prior to the implementation of new or revised quality standards, including those produced or sold by us, may not meet these standards, which could require us to discontinue marketing and/or recall such products from the market, either of which could adversely affect our business, financial position, and operating results. In addition, results of periodic testing we conduct on our products may indicate the presence of substances at levels above which are acceptable under FDA or other standards, which will require a recall of the product. For example, during the fourth quarter of 2019, testing of the API used in our ranitidine drug product, as well as testing of the drug product itself, indicated a level of a nitrosamine impurity called N-nitrosdimethylamine (“NDMA”) above acceptable thresholds. NDMA is classified as a probable human carcinogen. Appco Pharma, LLC, with whom we had partnered to develop and market the product, initiated a voluntary recall, and we elected to exit the market for Ranitidine in 2019. For a description of legal proceedings which are currently pending relating to ranitidine, see Note 12. Commitments and Contingencies, in the notes to the consolidated financial statements in Part II, Item 8 of this Annual Report on Form 10-K.
Another example of evolving standards occurred in December of 2021, when the FDA issued an information request to manufacturers of propranolol products, including Inderal LA (Propranolol ER) currently being marketed in the United States to evaluate their product for the presence and level of a nitrosamine impurity known as N-nitroso-propranolol (“NNP”), which is distinct from NDMA. Pfizer, Inc. and its affiliates (“Pfizer”), our contract manufacturer for both our Inderal LA brand product and our authorized generic product, Propranolol ER, initiated that evaluation and shared its analysis and test results with the Company in February 2022. Pfizer also manufactures and markets Inderal LA in Canada. On March 1, 2022, Pfizer announced that it was recalling all lots and strengths (60 mg, 80 mg, 120 mg, and 160 mg) of Inderal LA in the Canadian market after engagement with Health Canada. We are currently undertaking our own review and analysis of the nitrosamine impurity at issue, working with testing and toxicology experts, and are in active communication with the FDA on the appropriate acceptable daily intake for NNP, which has not been established. In the interim, we have halted further sales of the product to our trade customers. In March of 2022, we submitted our response to the FDA information request, including reference to both the evaluation performed by Pfizer and our review and evaluation to date performed with guidance from an independent third-party toxicologist. In addition, we requested a meeting with the FDA regarding the appropriate approach for the product in the U.S. Recently the FDA has responded and we anticipate a meeting in the near future.
The discussion above illustrates the potential risk of a recall of a product due to enhanced standards, at the initiation of the Company and/or the FDA. The loss of sales of this product would have an adverse effect on our results of operations, as revenues from Inderal LA and Propranolol ER are anticipated to contribute approximately 5% of our forecasted total 2022 ex-Cortrophin Net Revenues. In addition, Pfizer’s decision to withdraw the product in Canada creates uncertainties as to the future supply of our product from Pfizer which could have an adverse effect on our operating results if we are unable to supply the product pursuant to existing contracts with our customers.
29
We may become subject to federal and state false claims litigation brought by private individuals and the government.
We are subject to state and federal laws that govern the submission of claims for reimbursement. The Federal False Claims Act (“FFCA”), also known as Qui Tam, imposes civil liability and criminal fines on individuals or entities that knowingly submit, or cause to be submitted, false or fraudulent claims for payment to the government. Violations of the FFCA and other similar laws may result in criminal fines, imprisonment, and civil penalties for each false claim submitted and exclusion from federally funded health care programs, including Medicare and Medicaid. The FFCA also allows private individuals to bring a suit on behalf of the government against an individual or entity for violations of the FFCA. These suits, also known as Qui Tam actions, may be brought, with only a few exceptions, by any private citizen who has material information of a false claim that has not yet been previously disclosed. These suits have increased significantly in recent years because the FFCA allows an individual to share in any amounts paid to the federal government from a successful Qui Tam action. If our past or present operations are found to be in violation of any of such laws or other applicable governmental regulations, we may be subject to civil and criminal penalties, damages, fines, exclusion from federal health care programs, and/or the curtailment or restructuring of our operations, any of which could materially adversely affect our business, financial position, and operating results. Actions brought against ANI for violations of these laws, even if successfully defended, could also have a material adverse effect on our business, financial position, and operating results.
The use of legal, regulatory, and legislative strategies by competitors, both branded and generic, including "authorized generics," citizen’s petitions, and legislative proposals, may increase the costs to develop and market our generic products, could delay or prevent new product introductions, and could significantly reduce our profit potential. These factors could have a material adverse effect on our business, financial position, and operating results.
Our competitors, both branded and generic, often pursue legal, regulatory, and/or legislative strategies to prevent or delay competition from generic alternatives to branded products. These strategies include, but are not limited to:
| ● | entering into agreements whereby other generic companies will begin to market an authorized generic, a generic equivalent of a branded product, at the same time generic competition initially enters the market; |
| ● | launching a generic version of their own branded product at the same time generic competition initially enters the market; |
| ● | filing citizen petitions with the FDA or other regulatory bodies, including timing the filings so as to thwart generic competition by causing delays of generic product approvals; |
| ● | seeking to establish regulatory and legal obstacles that would make it more difficult to demonstrate bioequivalence or meet other approval requirements; |
| ● | initiating legislative and regulatory efforts to limit the substitution of generic versions of branded pharmaceuticals; |
| ● | filing suits for patent infringement that may delay regulatory approval of generic products; |
| ● | introducing "next-generation" products prior to the expiration of market exclusivity for the reference product, which often materially reduces the demand for the first generic product; |
| ● | obtaining extensions of market exclusivity by conducting clinical trials of branded drugs in pediatric populations or by other potential methods; |
| ● | persuading regulatory bodies to withdraw the approval of branded name drugs for which the patents are about to expire, thus allowing the branded company to obtain new patented products serving as substitutes for the products withdrawn; and |
| ● | seeking to obtain new patents on drugs for which patent protection is about to expire. |
If we cannot compete with such strategies, our business, financial position, and operating results could be adversely impacted.
30
If third-party payers deny coverage, substitute another company’s product for our product, or offer inadequate levels of reimbursement, we may not be able to market our products effectively or we may be required to offer our products at prices lower than anticipated.
Third-party payers are increasingly challenging the prices charged for medical products and services. For example, third-party payers may deny coverage, choose to provide coverage for a competitor’s bioequivalent product rather than our product, or offer limited reimbursement if they determine that a prescribed product has not received appropriate clearances from the FDA, is not used in accordance with cost-effective treatment methods as determined by the third-party payer, or is experimental, unnecessary, or inappropriate. Prices also could be driven down by health maintenance organizations that control or significantly influence purchases of healthcare services and products. If third-party payers deny coverage or limit reimbursement, we may not be able to market our products effectively or we may be required to offer our products at prices lower than anticipated.
We are subject to federal, state, and local laws and regulations, and complying with these may cause us to incur significant additional costs.
The pharmaceutical industry is subject to regulation by various federal authorities, including the FDA, the DEA, and state governmental authorities. Federal and state statutes and regulations govern or influence the testing, manufacturing, packing, labeling, storing, record keeping, safety, approval, advertising, promotion, sale, and distribution of our products. Noncompliance with applicable legal and regulatory requirements can have a broad range of consequences, including warning letters, fines, seizure of products, product recalls, total or partial suspension of production and distribution, refusal to approve NDAs or other applications or revocation of approvals previously granted, withdrawal of product from marketing, injunctions, withdrawal of licenses or registrations necessary to conduct business, disqualification from supply contracts with the government, civil penalties, debarment, and criminal prosecution.
All U.S. facilities where prescription drugs are manufactured, tested, packaged, stored, or distributed must comply with FDA current good manufacturing practices (“cGMPs”). All of our products are manufactured, tested, packaged, stored, and distributed according to cGMP regulations. The FDA performs periodic audits to ensure that our facilities remain in compliance with all applicable regulations. If it finds violations of cGMP, the FDA could make its concerns public and could impose sanctions including, among others, fines, product recalls, total or partial suspension of production and/or distribution, suspension of the FDA’s review of product applications, injunctions, and civil or criminal prosecution. If imposed, enforcement actions could have a material adverse effect on our business, financial position, and operating results. Under certain circumstances, the FDA also has the authority to revoke previously granted drug approvals. Although we have internal compliance programs in place that we believe are adequate, the FDA may conclude that these programs do not meet regulatory standards. If compliance is deemed deficient in any significant way, it could have a material adverse effect on our business.
The U.S. government has enacted the Federal Drug Supply Chain Security Act ("DSCSA") that requires development of an electronic pedigree to track and trace each prescription drug at the salable unit level through the distribution system, which will be effective incrementally over a 10-year period. All prescription pharmaceutical products distributed in the U.S. must be serialized with unique product identifiers. ANI started manufacturing serialization-compliant products in November 2018. The final requirement for tracking the products will commence on November 27, 2023. Compliance with DSCSA and future U.S. federal or state electronic pedigree requirements may increase the Company’s operational expenses and impose significant administrative burdens. In addition, if we are unable to comply with DSCSA as of the required dates, we could face penalties or be unable to sell our products.
Our research, product development, and manufacturing activities involve the controlled use of hazardous materials, and we may incur significant costs in complying with numerous laws and regulations. We are subject to laws and regulations enforced by the FDA, the DEA, and other regulatory statutes including the Occupational Safety and Health Act (“OSHA”), the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other current and potential federal, state, local, and foreign laws and regulations governing the use, manufacture, storage, handling, and disposal of our products, materials used to develop and manufacture such products, and resulting waste products.
31
We cannot completely eliminate the risk of contamination or injury, by accident or as the result of intentional acts, from these materials. In the event of an accident, we could be held liable for any damages that result, and any resulting liability could exceed our resources. We may also incur significant costs in complying with environmental laws and regulations in the future. We are also subject to laws generally applicable to businesses, including but not limited to, federal, state, and local regulations relating to wage and hour matters, employee classification, mandatory healthcare benefits, unlawful workplace discrimination, and whistle-blowing. Any actual or alleged failure to comply with any regulation applicable to our business or any whistle-blowing claim, even if without merit, could result in costly litigation, regulatory action or otherwise harm our business, financial position, and operating results.
Our operations in an international market subject us to additional regulatory oversight both in the international market and in the U.S., as well as, social, and political uncertainties, which could cause a material adverse effect on our business, financial position, and operating results.
We are subject to certain risks associated with having assets and operations located in a foreign jurisdiction, including our operations in Canada and India. Our Canadian operations are subject to regulation by Health Canada and other federal, provincial, and local regulatory authorities. Health Canada regulates the testing, manufacture, labeling, marketing, and sale of pharmaceutical products manufactured and distributed in Canada. Our operations in Canada and India may be adversely affected by general economic conditions and economic and fiscal policy, including changes in exchange rates and controls, interest rates and taxation policies, and increased government regulation, which could have a material adverse effect on our business, financial position, and operating results.
Continuing studies of our products could produce negative results, which could require us to implement risk management programs, or discontinue product marketing. In addition, ongoing post-approval drug safety surveillance of our products could result in the submission of adverse event reports to the FDA.
Studies of the proper utilization, safety, and efficacy of pharmaceutical products are being conducted by the industry, government agencies, and others on a continuous basis. Such studies, which increasingly employ sophisticated methods and techniques, can call into question the utilization, safety, and efficacy of current and previously marketed products, including those that we produce. In addition, we are required by the FDA to submit reports of adverse events involving the use of our products. In some cases, studies and safety surveillance programs have resulted, and in the future may result, in the one or more of the following:
These situations, should they occur with respect to any of our products, could have a material adverse effect on our business, financial position, and operating results.
Healthcare reform and changes in pharmaceutical pricing, reimbursement and coverage, by governmental authorities and third-party payors may materially affect our business, financial position and operating results.
In recent years, there have been numerous initiatives on the federal and state levels for comprehensive reforms affecting the payment for, the availability of, and reimbursement for healthcare services in the U.S. generally and prescription drug coverage, reimbursement and pricing specifically, and it is likely that federal and state legislatures will continue to advocate change to the healthcare system generally and to prescription drug coverage, reimbursement and pricing specifically.
At the federal level, the American Rescue Plan Act eliminated the cap on Medicaid Drug Rebate Program rebates beginning January 1, 2023. As such, we could end up owing additional rebates to state Medicaid programs related to utilization of our drug products negatively impacting profitability. States continue to look for ways to save on Medicaid spend specifically related to prescription drugs. As such, states are increasingly expanding or change supplemental rebates programs to secure additional rebates from manufacturers in exchange for drug coverage and to limit coverage of
32
certain drugs for certain Medicaid patients or to all Medicaid patients. To the extent the Centers for Medicare & Medicaid Services entertains waivers to federal requirements under the Medicaid program to allow states Medicaid programs such flexibility, coverage of and payment for our drugs utilized by Medicaid beneficiaries could be negatively impacted.
Significant developments that may adversely affect pricing in the United States include proposed drug pricing and Medicare reforms by Congress and regulatory changes to Medicare Part B (physician administered drugs) and Medicare Part D (prescription drug benefit) could financially impact us. On November 19, 2021, the U.S. House of Representatives passed the Build Back Better Act, which includes several provisions aimed at lowering prescription drug costs and reducing spending by the federal government and private payers by, among other things, allowing the U.S. federal government to negotiate prices for certain high-cost drugs covered under Medicare, imposing rebates on manufacturers of single-source drugs and biologics covered by Medicare Part B and nearly all drugs covered under Part D, if drug prices increase faster than the rate of inflation, based on the Consumer Price Index for All Urban Consumers (“CPI-U”). Build Back Better would also re-structure the Part D benefit and replace the existing Coverage Gap Discount Program with another manufacturer-imposed rebate or discount program, which could result in additional rebates to Medicare Part D plans in order to obtain Medicare Part D coverage. We are actively monitoring legislative developments to understand the likelihood of enactment and how such legislation would impact our business and operations, if enacted.
Certain U.S. states have implemented statutes aimed at prescription drug price transparency and some of those laws would permit state run boards or agencies to cap reimbursement for certain prescription drugs in the states. Such laws could negatively impact our financial performance and could result in us terminating distribution of certain products in certain states or regions.
Inflation could have a material adverse effect on our business, financial position, and operating results.
Inflationary pressures have begun to rapidly increase in the U.S. and key worldwide markets. The rate of inflation may significantly increase input costs for our products and, given the competitive nature of the generic markets in which we compete, we may not be able to pass those costs on to our generic customers.
Risks Related to Accounting, Tax, and SEC Rules and Regulations
We have increased exposure to tax liabilities, including foreign tax liabilities.
As a company based in the U.S. with subsidiaries in Canada and India, we are subject to, or potentially subject to, income taxes as well as non-income based taxes in these jurisdictions as well as the U.S. Significant judgment is required in determining our international provision for income taxes and other tax liabilities. Changes in tax laws or tax rulings may have a significantly adverse impact on our effective tax rate. In addition, we have potential tax exposures resulting from the varying application of statutes, regulations, and interpretations, which include exposures on intercompany terms of cross-border arrangements between our U.S. operations and our Canadian and Indian subsidiaries in relation to various aspects of our business, including research and development services, tech transfers, and contract manufacturing. Tax authorities in various jurisdictions may disagree with, and subsequently challenge, the amount of profits taxed in such jurisdictions; such challenges may result in increased tax liability, including accrued interest and penalties, which would cause our tax expense to increase and which could have a material adverse effect on our business, financial position and results of operations and our ability to satisfy our debt obligations.
Our expanded international operations from the Novitium acquisition increased our exposure to potential liability under anti-corruption, trade protection, tax, and other laws and regulations.
The Foreign Corrupt Practices Act and other anti-corruption laws and regulations (“Anti-Corruption Laws”) prohibit corrupt payments by our employees, vendors, or agents. From time to time, we receive inquiries from authorities in the U.S. and elsewhere about our business activities outside of the U.S. and our compliance with Anti-Corruption Laws. While we devote substantial resources to our global compliance programs and have implemented policies, training, and internal controls designed to reduce the risk of corrupt payments, our employees, vendors or agents may violate our policies and with the acquisition of Novitium, our expanded
33
international operations would significantly increase our exposure to potential liability. Our failure to comply with Anti-Corruption Laws could result in significant fines and penalties, criminal sanctions against us, our officers or our employees, prohibitions on the conduct of our business, and damage to our reputation. Operations outside of the U.S. may be affected by changes in trade production laws, policies, and measures, and other regulatory requirements affecting trade and investment.
We are also subject to Indian foreign tax regulations. Such regulations may not be clear, not consistently applied and subject to sudden change, particularly with regard to international transfer pricing. Our earnings could be reduced by the uncertain and changing nature of such tax regulations.
The global nature of Novitium’s operations (including those of its Indian subsidiary Novitium Labs Private Limited) will subject us to political and economic risks that could adversely affect our business, results of operations, or financial condition.
The risks presented by global operations include:
| • | limitations on ownership or participation in local enterprises; |
| • | price controls, exchange controls, and limitations on repatriation of earnings; |
| • | transportation delays and interruptions; |
| • | the application of additional legal, regulatory and taxation regimes to our operations; |
| • | political, social, and economic instability and disruptions in applicable regions; |
| • | acts of terrorism; |
| • | government embargoes or foreign trade restrictions; |
| • | imposition of duties and tariffs and other trade barriers; |
| • | import and export controls; |
| • | labor unrest and current and changing regulatory environments; |
| • | fluctuations in foreign current exchange and interest rates; |
| • | difficulties in staffing and managing multi-national operations; |
| • | limitations on our ability to enforce legal rights and remedies; and |
| • | the severity and duration of the COVID-19 pandemic and its impacts where we operate globally. |
If we are unable to successfully manage these and other risks associated with managing the expansion of our business to the jurisdictions in which Novitium operates, including India, the risks could have a material adverse effect on our business, results of operations, or financial condition.
Failure to comply with applicable transfer pricing and similar regulations could have a material adverse effect on our financial position and operating results.
We are subject to complex transfer pricing and other tax regulations in the United States, Canada, and India designed to ensure that appropriate levels of income are reported as earned and are taxed in the appropriate taxing jurisdictions. Although we believe that we are in substantial compliance with all applicable regulations and restrictions, we are subject to the risk that governmental authorities could audit our transfer pricing and related practices and assert that additional taxes are owed. In the event that the audits or assessments are concluded adversely against us, we may or may not be able to offset or mitigate the consolidated effect of any such assessments.
Changes in estimates regarding the fair value of goodwill or intangible assets may result in an adverse impact to our business, financial position, and operating results.
We test goodwill for impairment annually, or more frequently if changes in circumstances indicate that the carrying amount of goodwill might not be recoverable. Judgment is used in determining when these events and circumstances arise. We perform our review of goodwill based on our one reporting unit. If we determine that the carrying value of our assets may not be recoverable, we assess, using judgment and estimates, the fair value of our assets and to determine the
34
amount of any impairment loss, if any. Changes in judgments and estimates may result in the recognition of an impairment loss, which could have a material negative impact on our business, financial position, and operating results. While our testing in fiscal 2021 did not result in an impairment charge related to goodwill, there can be no assurances that our goodwill will not be impaired in the future.
Our material definite-lived intangible assets consist of ANDAs for previously marketed generic products, NDAs and product rights for our branded products, product rights related to certain generic products, and a non-compete agreement. These assets are being amortized over their useful lives of four to 10 years. For these definite-lived intangible assets, we perform an impairment analysis when events or circumstances indicate that the carrying value of the assets may not be recoverable. An impairment loss is recognized if, based on our impairment analysis, the carrying amount of the asset is not recoverable and its carrying amount exceeds its fair value. Any significant change in market conditions, estimates or judgments used to determine expected future cash flows that indicate a reduction in carrying value may give rise to impairment in the period that the change becomes known. An impairment charge could have a material negative impact on our business, financial position, and operating results. We recognized an impairment of $2.4 million in the year ended December 31, 2021, in relation to an ANDA asset, and there can be no assurances that our remaining intangible assets will not be impaired in the future.
Our management is required to devote substantial time to comply with public company regulations. If we are unable to comply with these regulations, investors could lose confidence in us, which could have a material adverse effect on our stock price, business, financial position, and operating results.
As a public company, we are required to comply with significant legal, accounting, and other requirements, and as a result, we incur significant regulatory compliance-related expenses. The Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act as well as rules implemented by the SEC and The Nasdaq Stock Market, impose various requirements on public companies, including those related to corporate governance practices. Our management and other personnel devote a substantial amount of time to these requirements. Some members of management do not have significant experience in addressing these requirements. Moreover, these rules and regulations have increased our legal and financial compliance costs relative to those of previous years and make some activities more time consuming and costly.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. In particular, we must perform system and process evaluation and testing of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. The Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) provides a framework for companies to assess and improve their internal control systems. Our compliance with these requirements has required that we incur substantial accounting and related expenses and expend significant management efforts. Moreover, if we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act, are unable to assert that our internal controls over financial reporting are effective, or identify deficiencies that are deemed to be material weaknesses, investors could lose confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline and we could be subject to sanctions or investigations by The Nasdaq Stock Market, the SEC, or other regulatory authorities. Any of these events could have a material adverse effect on our business, financial position, and operating results.
Our policies regarding returns, allowances and chargebacks, and marketing programs adopted by wholesalers may reduce revenues in future fiscal periods.
We, like other generic drug manufacturers, have agreements with customers allowing chargebacks, product returns, administrative fees, and other rebates. Under many of these arrangements, we may match lower prices offered to customers by competitors. If we choose to lower our prices, we generally give the customer a credit on the products that the customer is holding in inventory, which could reduce sales revenue for the period the credit is provided. Like our competitors, we also give credits for chargebacks to wholesalers with whom we have contracts for their sales to hospitals, group purchasing organizations, pharmacies, or other customers. A chargeback is the difference between the price at which we invoice the wholesaler and the price that the wholesaler’s end-customer pays for a product. Although we establish reserves based on prior experience and our best estimates of the impact that these policies may have in
35
subsequent periods, we cannot ensure that our reserves are adequate or that actual product returns, allowances, and chargebacks will not exceed our estimates.
Risks Related to our Debt
Making interest and principal payments under our Credit Facility consisting of $300.0 million term loan and a $40.0 million revolving credit facility, requires a significant amount of cash.
In connection with the completion of the Novitium acquisition, we entered into a new $300.0 million term loan and a $40.0 million revolving credit facility. The Credit Facility, which is secured by all our assets and the assets of our subsidiaries, was used to finance the cash consideration of the acquisition of Novitium and terminate and repay our previous senior credit facilities. In order to service the debt we incur under this facility, we will require a significant amount of cash. Our ability to make scheduled payments of principal and interest depends on our future performance, which is subject to economic, financial, competitive, and other factors beyond our control. Our business may not continue to generate cash flow from operations in the future sufficient to service our debt. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt, or obtaining additional debt or equity financing on terms that may not be favorable to us or available to us at all. Our ability to refinance any such debt will depend on the capital markets and our financial condition at that time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default under our current or future indebtedness. Any event of default or inability to otherwise satisfy our obligations could have a material adverse effect on our future operating results and financial condition.
Our Credit Agreement contains restrictive and financial covenants and if we are not in compliance with these covenants, our outstanding indebtedness under this facility could be accelerated and the lenders could terminate their commitments under the facility.
The Credit Agreement contains customary covenants that require maintenance of a leverage ratio at or below specified thresholds and restricts our ability to make certain distributions with respect to our capital stock, prepay other debt, make certain investments, encumber our assets, incur additional indebtedness, make capital expenditures, engage in certain business combinations, transfer, lease or dispose of our assets, alter the character of our business in any material respect or undertake various other corporate activities. Therefore, as a practical matter, these covenants restrict our ability to engage in or benefit from such activities. In addition, we pledged our assets in order to secure our repayment obligations under the New Credit Agreement. This pledge may reduce our operating flexibility because it restricts our ability to dispose of our assets or engage in other transactions that may be beneficial to us.
If we are unable to comply with the covenants in the Credit Agreement, we will be in default, which could result in the acceleration of our outstanding indebtedness and termination of funding commitments by the lenders. If such an acceleration occurs, we may not be able to repay our debt and we may not be able to borrow sufficient additional funds to refinance our debt, which would have a material adverse effect on our business, financial position, and operating results.
Changes in the method of determining London Interbank Offered Rate ("LIBOR"), or the replacement of LIBOR with an alternative reference rate, may adversely affect interest expense related to outstanding debt.
Amounts drawn under the New Credit Facility may bear interest rates in relation to LIBOR, depending on our selection. On July 27, 2017, the Financial Conduct Authority (“FCA”) in the United Kingdom announced that it would phase out LIBOR as a benchmark by the end of 2021. Subsequently, regulators have announced that most USD tenors of LIBOR, including LIBOR options of the New Credit Facility, will now cease on December 31, 2023. The U.S. Federal Reserve, in conjunction with the Alternative Reference Rates Committee, a steering committee comprised of large U.S. financial institutions, is recommending replacing U.S.-dollar LIBOR with the Secured Overnight Financing Rate (“SOFR”), a new index calculated by short-term repurchase agreements, backed by Treasury securities. At this time, it is not possible to predict the effect any discontinuance, modification or other reforms to LIBOR, or the establishment of alternative reference rates such as SOFR, or any other reference rate, will have on the Company or its borrowing costs.
36
Risks Related to our Common Stock
Our principal stockholders, directors, and executive officers own a significant percentage of our stock and will be able to exercise meaningful influence over our business.
Our current principal stockholders, directors, and executive officers beneficially own approximately 28% of our outstanding capital stock entitled to vote as of December 31, 2021. As a result, these stockholders, if acting together, would be able to influence or control matters requiring approval by our stockholders, including the election of directors and the approval of mergers, acquisitions, or other extraordinary transactions. They may also have interests that differ from stockholders generally and may vote in a way with which other stockholders disagree and which may be adverse to their interests. This concentration of ownership may have the effect of delaying, preventing, or deterring a change of control of ANI, could deprive stockholders of an opportunity to receive a premium for their common stock as part of a sale of ANI, and might ultimately affect the market price of our common stock.
Raising additional funds by issuing additional equity securities may cause dilution to our current stockholders. Raising additional funds by entering into additional credit or other borrowing facilities or issuing debt may subject us to covenants and other requirements that may restrict our operations.
We may seek to raise additional funds through the issuance of equity or equity-linked securities. If we were to raise funds through the issuance of equity or equity-linked securities, the percentage ownership of our stockholders could be diluted, potentially significantly, and these newly issued securities may have rights, preferences, or privileges senior to those of our existing stockholders. In addition, the issuance of any equity securities could be at a discount to the then-prevailing market price of our common stock.
If we require new debt financing, there is no assurance that such a transaction will be available on terms acceptable to us, or at all. In addition, we could be subject to onerous repayment terms or covenants that restrict our ability to operate our business and make distributions to our stockholders. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens, pay dividends, redeem our stock, or make investments. We can offer no assurance that any equity or debt financing transaction will be available on terms acceptable to us, or at all.
Provisions in our charter documents and Delaware law could discourage or prevent a takeover, even if such a transaction would be beneficial to our stockholders.
Provisions of our certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire ANI, even if doing so would be beneficial to our stockholders. These provisions include:
| ● | authorizing the issuance of “blank check” preferred shares that could be issued by our board of directors to increase the number of outstanding shares and thwart a takeover attempt; |
| ● | prohibiting cumulative voting in the election of directors, which would otherwise allow less than a majority of stockholders to elect director candidates; |
| ● | advance notice provisions in connection with stockholder proposals and director nominations that may prevent or hinder any attempt by our stockholders to bring business to be considered by our stockholders at a meeting or replace our board of directors; and |
| ● | as a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation law, which prevents certain stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of at least two-thirds of our outstanding common stock not held by such 15% or greater stockholder. |
Any provision of our certificate of incorporation and bylaws or Delaware law that has the effect of delaying, preventing, or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock.
37
General Risk Factors
We use a variety of estimates, judgments, and assumptions in preparing our consolidated financial statements. Estimates, judgments, and assumptions are inherently subject to change, and any such changes could result in corresponding changes to the amounts of assets, liabilities, revenues, expenses, and income. Any such changes could have a material adverse effect on our business, financial position, and operating results.
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) requires us to make estimates, judgments, and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the period. There are inherent uncertainties involved in estimates, judgments and assumptions, and any changes in estimates, judgments and assumptions used could have a material adverse effect on our business, financial position, and operating results.
In the consolidated financial statements included in the periodic reports filed with the SEC, estimates, judgments, and assumptions are used for, but not limited to, revenue recognition, allowance for credit losses, accruals for chargebacks, rebates, returns and other allowances, allowance for inventory obsolescence, stock-based compensation, valuation of financial instruments and intangible assets, allowances for contingencies and litigation, deferred tax assets and liabilities, deferred tax valuation allowance, contingent consideration, and the depreciable lives of fixed and intangible assets. Actual results could differ from those estimates. Estimates, judgments, and assumptions are inherently subject to change in the future, and any such changes could result in corresponding changes to the amounts of assets, liabilities, revenues, expenses, and income. Any such changes could have a material adverse effect on our business, financial position, and operating results.
The market price of our common stock has been volatile, and an investment in our common stock could decline in value.
The market price of our common stock has increased and decreased significantly and is likely to continue to fluctuate in the future. From time to time, the securities of small capitalization pharmaceutical companies, including ANI, experience significant market price fluctuations, often unrelated to these companies’ operating performance. In particular, the market price of our common stock may fluctuate significantly due to a variety of factors, including, but not limited to, regulatory or legal developments with respect to our industry, variations in our financial results or those of companies that are perceived to be similar to us, and rumors or new announcements by third parties, many of which are beyond our control and that may not be related to our operating performance.
In addition, the occurrence of any of the risks described in this report or in subsequent reports we file with the SEC could have a material adverse impact on the market price of our common stock. Securities class action litigation is sometimes brought against a company following periods of volatility in the market price of its securities or for other reasons. Securities litigation, whether with or without merit, could result in substantial costs and divert management’s attention and resources, which could harm our business, financial position, and operating results, as well as the market price of our common stock.
Shares of our common stock are relatively illiquid which may affect the market price of our common stock.
For the twelve months ended December 31, 2021, the average daily trading volume of our common stock on the NASDAQ Global Market was approximately 102,000 shares. Because of our relatively small public float, our common stock may be less liquid than the stock of companies with broader public ownership and trading of a relatively small volume of our common stock may have a greater impact on the market price for our shares than would be the case if our public float were larger.
Item 1B. Unresolved Staff Comments
None.
38
Item 2. Properties
Our corporate offices are located at 210 Main Street West, Baudette, Minnesota 56623. The facility, which we own, includes oral solid dose and liquid manufacturing and packaging, warehouse facilities, analytical, stability, and microbiological laboratory space, and employee office and mechanical space. We also own a manufacturing facility that includes oral solid dose manufacturing and packaging for pharmaceutical products that must be manufactured in a fully contained environment, warehouse facilities, and employee office and mechanical space. This facility is also located in Baudette, Minnesota. We also own a cold storage facility located in Baudette, Minnesota. In addition, we own a manufacturing facility located in Oakville, Ontario that includes oral solid dose, semi-solids, and non-sterile liquid manufacturing and packaging, warehouse facilities, analytical, stability, and microbiological laboratory space, and employee office and mechanical space. Through the acquisition of Novitium, we now also own a facility in East Windsor, New Jersey, which includes manufacturing, warehousing, laboratory, product development, and employee office space.
We lease spaces for finance employees in Minnetonka, Minnesota and Baudette, Minnesota, for warehouse and packaging activities in East Windsor, New Jersey and for research and development activities in Chennai, India. The leases will expire between 2022 and 2026.
We consider our leased and owned properties suitable and adequate for our current and foreseeable needs.
Item 3. Legal Proceedings
On March 24, 2021, Azurity Pharmaceuticals, Inc. (“Azurity”) filed a complaint in the United States District Court for the District of Minnesota against ANI Pharmaceuticals, Inc., asserting that ANI’s vancomycin hydrochloride oral solution drug product infringes U.S. Patent No. 10,688,046. The complaint sought injunctive relief, damages, including lost profits and/or royalty, treble damages, and attorneys’ fee and costs. On February 15, 2022, the Company entered into a settlement agreement with Azurity to resolve all claims related to this action. Under the terms of the agreement, Azurity granted ANI a non-exclusive, non-transferable, non-sublicensable, royalty-bearing license under its Patents to sell ANI product in the United States and dismissed the action with prejudice. In exchange, we paid Azurity $1.9 million of royalties from past sales and we will pay Azurity a royalty equal to 20% of gross margin of sales of the ANI product for a contractually defined term. We paid the settlement from cash on hand and the $1.9 million charge was recorded as cost of sales (excluding depreciation and amortization) on the consolidated statement of operations for the year ended December 31, 2021.
Our legal proceedings are discussed in Note 12. Commitments and Contingencies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock trades on the Nasdaq Global Market under the symbol “ANIP.”
39
Stockholder Information
As of March 8, 2022, there were approximately 218 shareholders of record of our common stock, which does not include stockholders that beneficially own shares held in a “nominee” or in “street” name, and six holders of record of Class C stock.
Dividends
We have never declared or paid cash dividends on our common stock. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Our shares of Series A convertible preferred stock (the “PIPE Shares”), accrue dividends at 6.50% per year on a cumulative basis, payable in cash or in-kind, and will also participate, on a pro-rata basis, in any dividends that may be declared with respect to our common stock. We currently intend to retain all remaining available funds and any future earnings to fund the development and growth of our business.
Recent Sales of Unregistered Securities
On November 19, 2021, we issued and sold to Ampersand 2020 Limited Partnership, an affiliate of Ampersand Capital Partners (the “PIPE Investor”), a related party, and the PIPE Investor purchased, 25,000 PIPE Shares, for a purchase price of $1,000 per share and an aggregate purchase price of $25.0 million, in a private placement issued in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended, and/or Regulation D promulgated thereunder.
Issuer Purchases of Equity Securities
None.
40
Performance Graph
The graph below compares the five-year cumulative total stockholder return on our common stock, the Nasdaq Stock Market (US) Index, and the Nasdaq Pharmaceuticals Index, assuming the investment of $100.00 on December 31, 2016, with dividends being reinvested. The stock price performance in the graph below is not necessarily indicative of future price performance.
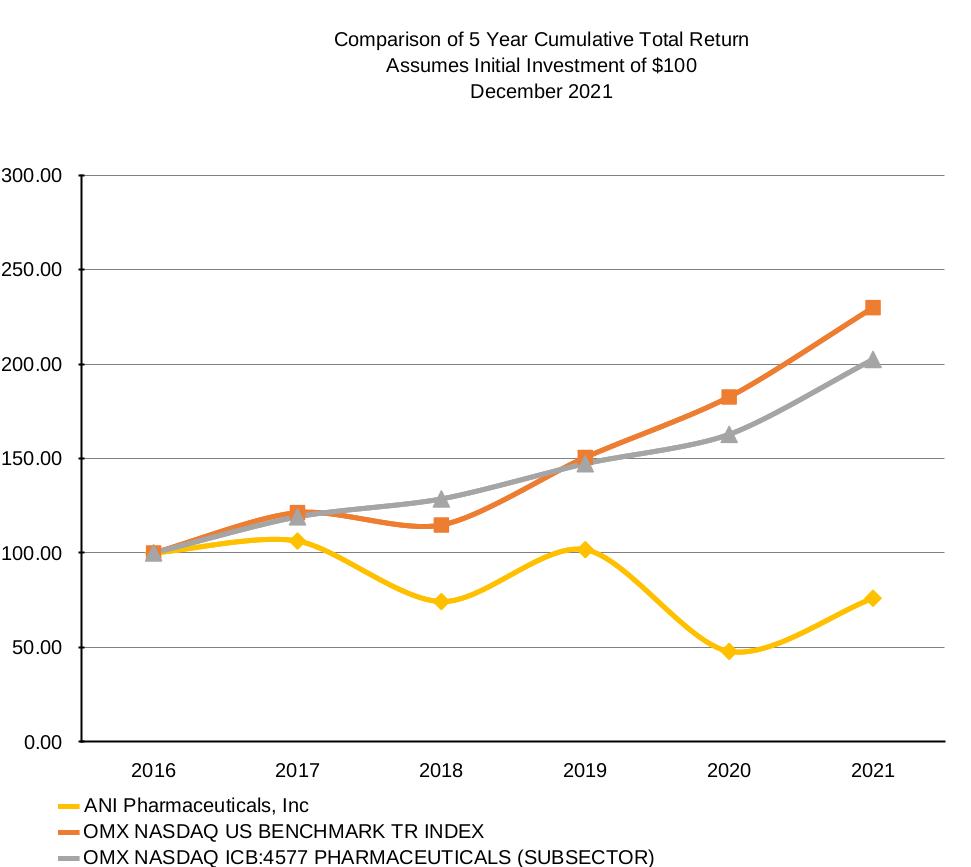
41
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
Please read the following discussion in conjunction with Item 1A. (“Risk Factors”) and our audited consolidated financial statements included elsewhere in this Annual Report on Form 10K. Some of the statements in the following discussion are forward-looking statements. See the discussion about forward-looking statements on page 1 of this Annual Report on Form 10-K.
This section of this Form 10-K generally discusses 2021 and 2020 items and year-to-year comparisons between 2021 and 2020. Discussions of 2019 items and year-to-year comparisons between 2020 and 2019 that are not included in this Form 10-K can be found in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in Part II, Item 7 of the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2020, filed with the SEC on March 11, 2021.
Executive Overview
ANI Pharmaceuticals, Inc. and its consolidated subsidiaries (together, “ANI,” the “Company,” “we,” “us,” or “our”) is a diversified bio-pharmaceutical company serving patients in need by developing, manufacturing, and marketing high quality branded and generic prescription pharmaceuticals, including for diseases with high unmet medical need. Our team is focused on delivering sustainable growth by building a successful Purified Cortrophin Gel franchise, strengthening our generics business with enhanced development capability, innovation in established brands and leveraging our North American manufacturing capabilities. Our four pharmaceutical manufacturing facilities, of which two are located in Baudette, Minnesota, one is located in East Windsor, New Jersey, and one is located in Oakville, Ontario, are together capable of producing oral solid dose products, as well as semi-solids, liquids and topicals, controlled substances, and potent products that must be manufactured in a fully-contained environment.
Strategy
Our objective is to build a sustainable and growing biopharmaceutical company serving patients in need and creating long-term value for our investors. Our growth strategy is driven by the following key pillars:
Building a successful Purified Cortrophin Gel franchise
We acquired the NDAs for Cortrophin gel and Cortrophin-Zinc in January 2016 and executed long-term supply agreements with a supplier of our primary raw material for corticotrophin active pharmaceutical ingredient (“API”), a supplier of corticotrophin API with whom we have advanced the manufacture of commercial scale batches of API, and a Cortrophin gel fill/finish contract manufacturer. During the second quarter of 2021, we submitted a Supplemental New Drug Application (“sNDA”) to the FDA.
On October 29, 2021, the FDA approved the Company’s sNDA for Purified Cortrophin™ Gel (Repository Corticotropin Injection USP) for the treatment of certain chronic autoimmune disorders, including acute exacerbations of multiple sclerosis (“MS”) and rheumatoid arthritis (“RA”), in addition to excess urinary protein due to nephrotic syndrome. Cortrophin Gel is an adrenocorticotropic hormone (“ACTH”), also known as purified corticotropin.
During 2021, we invested in leadership, expertise and infrastructure in the areas of commercialization of rare disease therapies and developed a launch strategy and commercial plan for this product. In the fourth quarter of 2021 and first quarter of 2022, we hired a significant number of new employees and assembled and trained our rare disease field force. On January 24, 2022, we announced the commercial launch of Cortrophin Gel in the U.S. As a result of the build out of our rare disease team, our expenditures in support of these efforts will materially increase in 2022 as compared to 2021.
Strengthening our generics business with enhanced research and development capability and increased focus on niche opportunities
43
We have grown our generics business through a combination of market share gains on existing products and new product launches. We have also successfully acquired numerous ANDAs through business and asset acquisitions, including, most recently, our acquisition of Novitium, including their portfolio of commercial and pipeline generic products, manufacturing and development facilities and expert workforce. We have begun to increase our focus on niche lower competition opportunities such as injectables, Paragraph IV, and Competitive Generic Therapy designation filings. Additionally, we will continue to seek opportunities to enhance our capabilities through strategic partnerships and acquisitions of assets and businesses.
Maximizing the value from our established brands through innovative “go-to-market” (“GTM”) strategies and continued programmatic acquisitions
We have acquired the New Drug Applications (“NDAs”) for and market Atacand, Atacand HCT, Arimidex, Casodex, Lithobid, Vancocin, Inderal LA, Inderal XL, InnoPran XL, Oxistat, Veregen, and Pandel. We are innovating in our GTM strategy through creative partnerships. In addition, we will continue to explore opportunities in acquiring new brands to grow our established brands portfolio.
Expansion of contract development and manufacturing organization (“CDMO”) business by leveraging our unique manufacturing capabilities
We built a CDMO business through our sites in Baudette and grew it through the acquisitions of Novitium and WellSpring Pharma Services Inc. (“ANI Canada”). Our North America based manufacturing and unique capabilities in high-potency, hormonal, steroid, and oncolytic products can be leveraged to expand our CDMO business.
The pillars of our strategy are enabled by an empowered, collaborative, and purposeful team with a high performance-orientation.
Product Development Considerations
We consider a variety of criteria in determining which products to develop, all of which influence the level of competition upon product launch. These criteria include:
| ● | Formulation Complexity. Our development and manufacturing capabilities enable us to manufacture pharmaceuticals that are difficult to produce, including highly potent, extended release, combination, and low dosage products. This ability to manufacture a variety of complex products is a competitive strength that we intend to leverage in selecting products to develop or manufacture. |
| ● | Patent Status. We seek to develop products whose branded bioequivalents do not have long-term patent protection or existing patent challenges. |
| ● | Market Size. When determining whether to develop or acquire an individual product, we review the current and expected market size for that product at launch, as well as forecasted price erosion upon conversion from branded to generic pricing. We endeavor to manufacture products with sufficient market size to enable us to enter the market with a strong likelihood of being able to price our products both competitively and at a profit. |
| ● | Profit Potential. We research the availability and cost of active pharmaceutical ingredients in determining which products to develop or acquire. In determining the potential profit of a product, we forecast our anticipated market share, pricing, including the expected price erosion caused by competition from other generic manufacturers, and the estimated cost to manufacture the products. |
| ● | Manufacturing. We generally seek to develop and manufacture products at our own manufacturing plants in order to optimize the utilization of our facilities, ensure quality control in our products, and to more closely control the economic inputs and outputs of our products. |
44
| ● | Competition. When determining whether to develop or acquire a product, we research existing and expected competition. We seek to develop products for which we can obtain sufficient market share and may decline to develop a product if we anticipate significant competition. Our specialized manufacturing facilities provide a means of entering niche markets, such as hormone therapies, in which fewer generic companies are able to compete. |
Fiscal 2021 Developments
Business Acquisition and Financing Activity
On November 19, 2021, we completed our previously announced acquisition of Novitium pursuant to the Merger Agreement for cash consideration of $89.5 million in cash (subject to various adjustments pursuant to the merger agreement), 2,466,654 restricted shares of ANI common stock, and up to $46.5 million in contingent future earn-out payments. The contingent consideration is based on the achievement of certain milestones, including milestones on gross profit of Novitium portfolio products over a 24-month period, regulatory filings completed during this 24-month period, and a percentage of net profits on certain products that are launched in the future. The cash consideration was financed via proceeds from the Credit Facility (as defined below) and proceeds from the PIPE Investment (as defined below). This acquisition is being accounted for as a business combination.
We acquired Novitium due to its proven track record of being a research and development growth engine capable of fueling sustainable growth, to expand our research and development pipeline via niche opportunities, to enhance our contract development and manufacturing organization (“CDMO”) business and U.S. based manufacturing capacity, and to diversify our revenue base.
In connection with our acquisition of Novitium, we entered into employment agreements with the two executives and founders of Novitium, Muthusamy Shanmugam and Chad Gassert. Both will serve as executive officers of the Company and Mr. Shanmugam was also appointed to the board of directors. Mr. Shanmugam holds interests in Scitus Pharma Services (“Scitus”), which provides clinical research services to Novitium, SS Pharma LLC (“SS Pharma”), which acquires and supplies API to Novitium, Esjay Pharma LLC (“Esjay”), which provides research and development and facilities consulting services, and Nuray Chemical Private Limited (“Nuray”), which manufactures and supplies API to Novitium. Mr. Gassert holds an interest in Scitus. See Note 14, Related Parties, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional discussion.
Also on November 19, 2021, we, as borrower, entered into a credit agreement (the “Credit Agreement”) with Truist Bank, as Administrative Agent, and the other parties thereto, which provides for credit facilities consisting of (i) a senior secured term loan facility in an aggregate principal amount of $300.0 million (the “Term Facility”) and (ii) a senior secured revolving credit facility in an aggregate commitment amount of $40.0 million, which may be used for revolving credit loans, swingline loans and letters of credit (the “Revolving Facility,” and together with the Term Facility, the “Credit Facility”). The Term Facility proceeds were used to finance the cash portion of the consideration under the merger agreement between ANI and Novitium, fully repay and terminate our existing Amended and Restated Credit Agreement (“Prior Credit Facility”), and pay fees, costs and expenses incurred in connection with the merger.
The Credit Facility is secured by substantially all of the personal property and certain material real property owned by us and our wholly-owned domestic subsidiaries, and obligations under the Credit Facility are guaranteed by certain of our wholly-owned domestic subsidiaries.
Concurrently with the execution of the Merger Agreement on March 8, 2021, we entered into an Equity Commitment and Investment Agreement with Ampersand 2020 Limited Partnership, an affiliate of Ampersand Capital Partners (the “PIPE Investor”), pursuant to which, on November 19, 2021, we issued and sold to the PIPE Investor, and the PIPE Investor purchased, 25,000 PIPE Shares, for a purchase price of $1,000 per share and an aggregate purchase price of $25.0 million, in a private placement (the “PIPE Investment”) issued in reliance on the exemption from registration provided by Section (4(a)(2) of the Securities Act of 1933, as amended and/or Regulation D promulgated thereunder. Our Chairman of the Board of Directors is an operating partner of Ampersand Capital Partners, an affiliate of Ampersand 2020 Limited Partnership.
45
As part of the Federal Trade Commission (“FTC”) conditions on the closing of the acquisition of Novitium, we agreed to divest a currently marketed product and rights to another product under development to an unrelated third party. The disposition of these products is immaterial to our results of operations.
For more information about the Novitium acquisition transaction, please see our Form 8-K filed with the SEC on November 26, 2021.
Asset Acquisitions
On April 1, 2021, we acquired the NDAs for Oxistat®, Veregen®, and Pandel® and the ANDA for Apexicon® from Sandoz Inc. for total consideration of $20.7 million. The acquisition was funded through a $24.0 million borrowing under the revolving facility portion (the “Revolver”) of our Prior Credit Facility.
Product Launches
Refer to our website at www.anipharmaceuticals.com for information on the products, including indications/treatments.
Purified Cortrophin Gel Approval and Launch
On October 29, 2021, the FDA approved the Company’s sNDA for Purified Cortrophin™ Gel (Repository Corticotropin Injection USP) for the treatment of certain chronic autoimmune disorders, including acute exacerbations of multiple sclerosis (“MS”) and rheumatoid arthritis (“RA”), in addition to excess urinary protein due to nephrotic syndrome. Cortrophin Gel is an adrenocorticotropic hormone (“ACTH”), also known as purified corticotropin.
During 2021, we invested in leadership, expertise and infrastructure in the areas of commercialization of rare disease therapies and developed a launch strategy and commercial plan for this product. In the fourth quarter of 2021 and first quarter of 2022, we hired a significant number of new employees and assembled and trained our rare disease field force. As a result of the build out of our rare disease team, our expenditures in support of these efforts will materially increase in 2022 as compared to 2021.
Purified Cortrophin Gel became available to our customers in late 2021, and we recognized an immaterial amount of revenues during the year ended December 31, 2021. On January 24, 2022, we announced the full-scale U.S. commercial availability and launch of Purified Cortrophin Gel.
Equity Financing
In November 2021, through a public offering, we completed the issuance and sale of 1,500,000 shares of ANI common stock, resulting in net proceeds after issuance costs of $69.7 million. The proceeds will be used to fund our Purified Cortrophin Gel commercialization efforts, including sales and marketing and consulting expenses related thereto, and for general corporate purposes.
COVID-19 Impact
We continue to closely monitor the impact of the novel coronavirus (“COVID-19”) pandemic on our business and the geographic regions where we operate. Per IQVIA/IMS data, total market generic and brand prescriptions continued to be depressed during 2021. During this period, and most significantly in the first quarter of 2021, our revenues were negatively impacted by the pandemic, as subsequent waves and variants of the virus impacted patient and customer behavior. IQVIA/IMS data indicates that total market generic and brand prescriptions increased sequentially during each of the second, third, and fourth quarterly periods of 2021 and increased against the comparable 2020 quarterly periods, and prescription levels appear to be nearing or have returned to pre-pandemic levels by the end of 2021. We have not experienced a significant impact to our manufacturing operations; however, we continued to see disruptions to our supply chain from the COVID-19 pandemic during 2021, including significant lead times for purchases of materials. Our
46
manufacturing facilities have remained open throughout the pandemic and have operated in accordance with local, state and national safety guidelines. The pandemic has not impacted our access to capital and has not significantly impacted our use of funds, including but not limited to capital expenditures, spend on research and development activities and business development opportunities.
We are unable to predict the impact that the COVID-19 pandemic will continue to have on our future financial condition, results of operations and cash flows due to numerous uncertainties, including the continued duration of the pandemic, the appearance of additional variants of the virus, the level of success of continued actions taken to contain the pandemic or mitigate its impact, and the direct and indirect economic effects of the pandemic and containment measures, among others.
General
Impacts to our 2021 results of operations, including to net revenues, operating expenses, interest and other expense, net, and income taxes are described below. Our 2021 results of operations were impacted by the November 19, 2021 acquisition of Novitium and related activity subsequent to that date. The acquisition will provide additional revenues and we will incur increased costs, including but not limited to the amortization of intangible assets acquired, other operating costs, and increased interest costs on borrowings used to finance the transaction. During the period between the acquisition date and December 31, 2021, Novitium operations generated $7.7 million in net revenues.
The following table summarizes our results of operations for the periods indicated:
Year Ended | ||||||
December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Net revenues | $ | 216,136 | $ | 208,475 | ||
Operating expenses |
|
|
| |||
Cost of sales (exclusive of depreciation and amortization) |
| 100,610 |
| 87,157 | ||
Research and development |
| 11,369 |
| 16,001 | ||
Selling, general, and administrative |
| 84,294 |
| 64,986 | ||
Depreciation and amortization |
| 47,252 |
| 44,638 | ||
Contingent consideration fair value adjustment | 500 | — | ||||
Legal settlement expense | 8,750 | — | ||||
Purified Cortrophin Gel pre-launch charges |
| 780 |
| 11,263 | ||
Intangible asset impairment charge | 2,374 | 446 | ||||
Operating loss |
| (39,793) |
| (16,016) | ||
Interest expense, net |
| (11,922) |
| (9,452) | ||
Other expense, net |
| (4,343) |
| (494) | ||
Loss before benefit for income taxes |
| (56,058) |
| (25,962) | ||
Benefit for income taxes |
| 13,455 |
| 3,414 | ||
Net loss | $ | (42,603) | $ | (22,548) | ||
47
The following table sets forth, for the periods indicated, items in our consolidated statements of operations as a percentage of net revenues.
Year Ended |
| ||||
December 31, |
| ||||
| 2021 |
| 2020 |
| |
Net revenues |
| 100.0 | % | 100.0 | % |
Operating expenses |
|
|
|
| |
Cost of sales (exclusive of depreciation and amortization) |
| 46.5 | % | 41.8 | % |
Research and development |
| 5.3 | % | 7.7 | % |
Selling, general, and administrative |
| 39.0 | % | 31.2 | % |
Depreciation and amortization |
| 21.9 | % | 21.4 | % |
Contingent consideration fair value adjustment | 0.2 | % | — | % | |
Legal settlement expense | 4.0 | % | — | % | |
Purified Cortrophin Gel pre-launch charges |
| 0.4 | % | 5.4 | % |
Intangible asset impairment charge |
| 1.1 | % | 0.2 | % |
Operating loss |
| (18.4) | % | (7.7) | % |
Interest expense, net |
| (5.5) | % | (4.5) | % |
Other expense, net |
| (2.0) | % | (0.2) | % |
Loss before benefit for income taxes |
| (25.9) | % | (12.4) | % |
Benefit for income taxes |
| 6.2 | % | 1.6 | % |
Net loss |
| (19.7) | % | (10.8) | % |
Results of Operations for the Years Ended December 31, 2021 and 2020
Net Revenues
Year Ended December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Change |
| % Change |
| |||
Generic pharmaceutical products | $ | 143,571 | $ | 147,257 | $ | (3,686) |
| (2.5) | % | |||
Branded pharmaceutical products |
| 47,561 |
| 47,960 |
| (399) |
| (0.8) | % | |||
Contract manufacturing |
| 10,042 |
| 9,221 |
| 821 |
| 8.9 | % | |||
Royalty and other income |
| 14,962 |
| 4,037 |
| 10,925 |
| 270.6 | % | |||
Total net revenues | $ | 216,136 | $ | 208,475 | $ | 7,661 |
| 3.7 | % | |||
We derive substantially all of our revenues from sales of generic and branded pharmaceutical products, contract manufacturing, and contract services, which include product development services, laboratory services, and royalties on net sales of certain products. Many of our branded products face competition from generic products and we expect them to continue to face competition from generic products in the future. Our generic products face competition from other generic products and we expect them to continue to face competition in the future. The primary means of competition among generic manufacturers are pricing, contract terms, service levels, and reliability. Increased competition generally results in decreased average selling prices of generic and brand products over time. In addition, due to strategic partnerships between wholesalers and pharmacy chains, we have experienced, and expect to continue to experience, increases in net sales to the wholesalers, with corresponding decreases in net sales to the pharmacy chains.
Net revenues for the year ended December 31, 2021 were $216.1 million compared to $208.5 million for the same period in 2020, an increase of $7.7 million, or 3.7%, primarily as a result of the following factors:
| ● | Net revenues for generic pharmaceutical products were $143.6 million during the year ended December 31, 2021, a decrease of 2.5% compared to $147.3 million for the same period in 2020. From a product perspective, the net decrease was driven by declines in sales of Vancomycin, Methazolamide, Erythromycin Ethylsuccinate (“EES”), Miglustat, Penicillamine, and Tolterodine, and tempered by increased revenues from sales of Flecainide, the second quarter 2021 launch of Nicardipine, and the third quarter 2021 launches of Nebivolol and Tranexamic Acid, and the sales of generic products acquired in the Novitium acquisition from November 19, 2021 through the year ended December 31, 2021. The decrease in net generic revenues was principally due to |
48
| lower average selling prices among generic products, which was tempered by an increase in volumes of generic products other than those mentioned above. |
During the year ended December 31, 2020, the overall market for and sales of our generic products were negatively impacted by the COVID-19 pandemic, as mitigation measures and other related actions suppressed prescription levels during the year. During the year ended December 31, 2021, generic prescription levels continued to be suppressed when compared to pre-pandemic levels, most significantly during the three months ended March 31, 2021, and the revenues for many of our generic pharmaceutical products continued to be negatively impacted. Per IQVIA/IMS data, total generic market prescriptions increased sequentially during the second, third, and fourth quarterly periods in 2021 and appear to be nearing pre-pandemic levels.
| ● | Net revenues for branded pharmaceutical products were $47.6 million during the year ended December 31, 2021, a decrease of 0.8% compared to $48.0 million for the same period in 2020. From a product perspective, the net decrease was driven by lower unit sales of Inderal XL and Arimidex and decreased units and revenues of Atacand. These decreases were tempered by the launch of the products acquired in the Sandoz, Inc. asset acquisition on April 1, 2021 and increased unit sales and revenues of Casodex. Net brand revenues in 2021 were negatively impacted by a shift in mix towards products with lower average selling prices, tempered by an increase in overall volumes. |
During the year ended December 31, 2020, the overall market for, and sales of our brand products were negatively impacted by the COVID-19 pandemic, as mitigation measures and other related actions suppressed prescription levels throughout the year. These actions resulted in suppressed brand prescriptions during the year ended December 31, 2020. As of the end of the 2021 fiscal year, brand prescription levels appeared to have returned to pre-pandemic levels.
| ● | Contract manufacturing revenues were $10.0 million during the year ended December 31, 2021, an increase of 8.9% compared to $9.2 million for the same period in 2020, due to an increase in the volume of orders, including the impact of Novitium contract manufacturing orders during the period from November 19, 2021 and December 31, 2021. |
| ● | Royalty and other were $15.0 million during the year ended December 31, 2021, an increase of $10.9 million from $4.0 million for the same period in 2020, primarily due to the recognition of the final royalty of $11.2 million under the Kite Pharma, Inc. license agreement (Yescarta®) pursuant to the Tripartite Agreement in the first quarter 2021. |
Cost of Sales (Excluding Depreciation and Amortization)
Year Ended December 31, |
| |||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Change |
| % Change |
| |||
Cost of sales (excl. depreciation and amortization) | $ | 100,610 | $ | 87,157 | $ | 13,453 |
| 15.4 | % | |||
Cost of sales consists of direct labor, including manufacturing and packaging, active and inactive pharmaceutical ingredients, freight costs, packaging components, and royalties related to profit-sharing arrangements. Cost of sales does not include depreciation and amortization expense, which is reported as a separate component of operating expenses on our consolidated statements of operations.
For the year ended December 31, 2021, cost of sales increased to $100.6 million from $87.2 million for the same period in 2020, an increase of $13.5 million or 15.4%. The increase is primarily due to increased volumes of generic products, including increases related to activity of Novitium subsequent to our acquisition, a $3.2 million increase in costs representing the excess of fair value over cost for inventory acquired in asset acquisitions and a business combination, $1.9 million in a non-recurring royalty settlement, and $1.5 million of increased freight charges during the year ended December 31, 2021. The increase was tempered by $3.5 million of lower costs related to a current period decrease in sales of products subject to profit sharing arrangements. During the year ended December 31, 2021, we
49
incurred $7.5 million in cost of sales representing the excess of fair value over cost for inventory acquired in the Sandoz, Inc. asset acquisition and Novitium business combination and subsequently sold during the period, compared to $4.3 million during the year ended December 31, 2020, related to the Amerigen asset acquisition.
Cost of sales as a percentage of net revenues, exclusive of the impacts related to excess of fair value over the cost of inventory sold during the period, increased to 43.1% during the year ended December 31, 2021, from 39.7% during the same period in 2020, primarily as a result of increased volumes in a period of declining average selling prices across generic products and a shift in mix towards brand products with lower average selling prices, as well as a $1.9 million non-recurring royalty settlement and a $1.5 million increase in freight expenses. The negative impacts were significantly tempered by $11.2 million of royalty revenue in the first quarter 2021 with no associated cost of sales.
During the year ended December 31, 2021, no single vendor represented at least 10% of inventory purchases. In the year ended December 31, 2020, we purchased 10% of our inventory from one supplier.
Other Operating Expenses
Year Ended December 31, | ||||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Change |
| % Change |
| |||
Research and development | $ | 11,369 | $ | 16,001 | $ | (4,632) |
| (28.9) | % | |||
Selling, general, and administrative |
| 84,294 |
| 64,986 |
| 19,308 |
| 29.7 | % | |||
Depreciation and amortization |
| 47,252 |
| 44,638 |
| 2,614 |
| 5.9 | % | |||
Contingent consideration fair value adjustment | 500 | — | 500 | NM | (1) | |||||||
Legal settlement expense | 8,750 | — | 8,750 | NM | (1) | |||||||
Purified Cortrophin Gel pre-launch charges |
| 780 |
| 11,263 |
| (10,483) |
| (93.1) | % | |||
Intangible asset impairment charge | 2,374 | 446 | 1,928 | 432.3 | % | |||||||
Total other operating expenses | $ | 155,319 | $ | 137,334 | $ | 17,985 |
| 13.1 | % | |||
| (1) | Not meaningful |
Other operating expenses consist of research and development costs, selling, general, and administrative expenses, depreciation and amortization, contingent consideration fair value adjustment, legal settlement expense, Purified Cortrophin Gel pre-launch charges, and intangible asset impairment charges.
For the year ended December 31, 2021, other operating expenses increased to $155.3 million from $137.3 million for the same period in 2020, an increase of $18.0 million, or 13.1%, primarily as a result of the following factors:
| ● | Research and development expenses decreased from $16.0 million to $11.4 million, a decrease of 28.9%, primarily due to the non-recurrence of the $3.8 million in-process research and development expense from the Amerigen Pharmaceuticals, Ltd. acquisition in the first quarter 2020. The decrease was tempered by increases related to the Novitium activities subsequent to our acquisition. |
| ● | Selling, general, and administrative expenses increased from $65.0 million to $84.3 million, an increase of 29.7%, primarily due to the $9.4 million of transaction expenses related to the Novitium acquisition and $14.0 million in pre-launch sales and marketing expenses related to Cortrophin commercialization activities incurred in the year ended December 31, 2021. In 2020, there were no comparable costs. Increased costs were also incurred related to employee compensation, legal, insurance, and other professional fees, in part related to Novitium activities subsequent to the acquisition. These increases were offset by the non-recurrence of $6.5 million of termination benefit expenses related to the departure of our former President and CEO and non-recurrence of other recruitment and related legal charges associated with our CEO search in the second quarter of 2020. |
| ● | Depreciation and amortization expense was $47.3 million for the year ended December 31, 2021, compared to $44.6 million for the year ended December 31, 2020. The increase is primarily due to the amortization of intangible assets acquired in the Novitium acquisition. |
50
| ● | As described in Note 8, Fair Value Disclosures, in the notes to the consolidated financial statements included in Part II, Item 8. of this Annual Report on Form 10-K, we recognized a contingent consideration fair value adjustment of $0.5 million in the year ended December 31, 2021. No contingent consideration fair value adjustment was recognized in the year ended December 31, 2020. |
| ● | As described in Note 12, Commitments and Contingencies, in the notes to the consolidated financial statements included in Part II, Item 8. of this Annual Report on Form 10-K, we recognized legal settlement expense of $8.8 million in the year ended December 31, 2021, principally related to settlement of the Arbor matter. No legal settlement expenses were recognized in the year ended December 31, 2020. |
| ● | As described in Note 13, Purified Cortrophin Gel Pre-Launch Charges, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, we recognized Cortrophin pre-launch charges related to purchases of materials of $0.8 million in the year ended December 31, 2021. We recognized Cortrophin pre-launch charges related to purchases of materials of $11.3 million in the year ended December 31, 2020. The decrease is due to sufficient levels of materials acquired in prior periods. |
| ● | We recognized an impairment of $2.4 million in the year ended December 31, 2021, in relation to an ANDA asset. We recognized an impairment charge of $0.4 million in relation to a marketing and distribution right intangible asset during the year ended December 31, 2020. |
Other Expense, net
Year Ended December 31, | ||||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Change |
| % Change |
| |||
Interest expense, net | $ | (11,922) | $ | (9,452) | $ | (2,470) |
| 26.1 | % | |||
Other expense, net |
| (4,343) |
| (494) |
| (3,849) |
| 779.1 | % | |||
Total other expense, net | $ | (16,265) | $ | (9,946) | $ | (6,319) |
| 63.5 | % | |||
For the year ended December 31, 2021, we recognized other expense, net of $16.3 million versus other expense, net of $9.9 million for the same period in 2020, an increase of $6.3 million. Interest expense, net for 2021 and 2020 consists primarily of interest expense on our Term Loan, DDTL, and Revolver under our Prior Credit Facility, and interest expense on our new Term Facility subsequent to the termination of our Prior Credit Facility and entry into new Credit Facility on November 19, 2021. The increase in interest expense in the year ended December 31, 2021 is due to $24.0 million of additional borrowings under our Revolver of the Prior Credit Facility in April 2021 and the increased borrowings and borrowing rate on our new $300.0 million Term Facility draw on November 19, 2021. For the year ended December 31, 2021, other expense, net primarily consisted of $4.2 million ticking fee expense related to our Credit Facility that was syndicated on May 24, 2021, a $1.5 million loss on the extinguishment of debt related to our Prior Credit Facility, and $1.8 million in net gains on the sale of ANDAs. None of these items occurred in the comparable period of 2020. For the year ended December 31, 2021 and 2020, there was $0.1 million of interest capitalized into construction in progress.
Benefit for Income Taxes
Year Ended December 31, | ||||||||||||
(in thousands) |
| 2021 |
| 2020 |
| Change |
| % Change |
| |||
Benefit for income taxes | $ | 13,455 | $ | 3,414 | $ | 10,041 |
| 294.1 | % | |||
Our provision for income taxes consists of current and deferred components, which include changes in our deferred tax assets, our deferred tax liabilities, and our valuation allowance. We measure our deferred tax assets and liabilities using the tax rates that we believe will apply in the years in which the temporary differences are expected to be recovered or paid. See Note 11. Income Taxes, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for further information.
51
For the year ended December 31, 2021, we recognized an income tax benefit of $13.5 million, an effective benefit rate of 24.0% of consolidated pre-tax losses reported in the period. Our effective tax rate for 2021 was impacted by changes in state tax rates due to our increased presence in certain states, certain non-deductible expenses, and the impact of current period stock-based compensation, among other items.
For the year ended December 31, 2020, we recognized an income tax benefit of $3.4 million, an effective benefit rate of 13.1% of consolidated pre-tax losses reported in the period. Our effective tax rate for 2020 was impacted by changes in state tax rates due to our changing presence in certain states, certain non-deductible expenses, and the impact of current period stock-based compensation, among other items.
Liquidity and Capital Resources
The following table highlights selected liquidity and working capital information from our consolidated balance sheets.
December 31, | December 31, | |||||
(in thousands) |
| 2021 |
| 2020 | ||
Cash and cash equivalents | $ | 100,300 | $ | 7,864 | ||
Accounts receivable, net |
| 128,526 |
| 95,793 | ||
Inventories, net |
| 81,693 |
| 60,803 | ||
Prepaid income taxes |
| 3,667 |
| — | ||
Prepaid expenses and other current assets |
| 7,589 |
| 5,861 | ||
Total current assets | $ | 321,775 | $ | 170,321 | ||
Current debt, net of deferred financing costs | $ | 850 | $ | 13,243 | ||
Accounts payable |
| 22,967 |
| 11,261 | ||
Accrued expenses and other |
| 7,563 |
| 2,456 | ||
Accrued royalties |
| 6,225 |
| 6,407 | ||
Accrued compensation and related expenses |
| 8,522 |
| 6,231 | ||
Current income taxes payable, net |
| — |
| 3,906 | ||
Accrued government rebates |
| 5,492 |
| 7,826 | ||
Returned goods reserve |
| 35,831 |
| 27,155 | ||
Deferred revenue |
| 87 |
| 80 | ||
Total current liabilities | $ | 87,537 | $ | 78,565 | ||
On December 31, 2021, we had $100.3 million in unrestricted cash and cash equivalents. On December 31, 2020, we had $7.9 million in unrestricted cash and cash equivalents. During 2021, we began investing in leadership, expertise, and infrastructure in the areas of commercialization of rare disease therapies and have developed a commercial plan for our Cortrophin Gel product. We anticipate that our expenditures in support of these efforts will materially increase in 2022 as we increase headcount and incur other costs associated with the launch.
We financed the acquisition of Novitium in part with borrowings under the Credit Facility described below under “Sources and Uses of Cash – Debt Financing,” and by a $25.0 million PIPE Investment by Ampersand 2020 Limited Partnership (“Ampersand”).
In January 2020, we acquired the U.S. portfolio of 23 generic products and certain commercial and development inventory and materials from Amerigen Pharmaceuticals, Ltd., for which we have used $57.4 million in cash and could make future payments of up to $25.0 million in contingent profit share payments over the next two years. The contingent payments are earned if annual gross profit exceeds a minimum threshold and are earned on a subset of the acquired products. No payment was due to Amerigen for the fiscal years ended December 31, 2021 or 2020. The transaction was funded from cash on hand and $15.0 million of borrowings from our Revolver, of which $7.5 million was repaid in the second quarter 2020. In July 2020, we acquired an ANDA and certain inventories from a private company for total consideration of $4.3 million. The transaction was funded using cash on hand. During 2020, we incurred expenses of $11.3 million related to purchases of Cortrophin pre-launch inventory.
52
We are focused on expanding our business and product pipeline through collaborations, and also through acquisitions of products and companies. We are continually evaluating potential asset acquisitions and business combinations. To finance such acquisitions, we might raise additional equity capital, incur additional debt, or both.
Our working capital ratio, defined as total current assets divided by total current liabilities, is 3.7 as of December 31, 2021. We believe that our financial resources, consisting of current working capital, anticipated future operating revenue and corresponding collections from customers, and our Credit Facility, under which $40.0 million remains available for borrowing as of December 31, 2021, will be sufficient to enable us to meet our working capital requirements and debt obligations for at least the next 12 months. If our assumptions underlying estimated revenue and expenses are wrong, or if our cash requirements change materially as a result of shifts in our business or strategy, we could require additional financing. If we are not profitable or do not generate cash from operations as anticipated and additional capital is needed to support operations, we may be unable to obtain such financing, or obtain it on favorable terms, in which case we may be required to curtail development of new products, limit expansion of operations, or accept financing terms that are not as attractive as desired.
Consolidation among wholesale distributors, chain drug stores, and group purchasing organizations has resulted in a smaller number of companies each controlling a larger share of pharmaceutical distribution channels. Our net revenues were concentrated among three customers representing 29%, 23%, and 16% of net revenues during the year ended December 31, 2021. As of December 31, 2021 accounts receivable from these three customers totaled approximately 92% of accounts receivable, net. As a result, negotiated payment terms with these customers have a material impact on our liquidity and working capital.
None of our products accounted for 10% or more of our net revenues in 2021 or 2020.
Sources and Uses of Cash
Debt Financing
On November 19, 2021, the Company, as borrower, entered into a credit agreement (the “Credit Agreement”) with Truist Bank and other lenders, which provides for credit facilities consisting of (i) a senior secured term loan facility in an aggregate principal amount of $300.0 million (the “Term Facility”) and (ii) a senior secured revolving credit facility in an aggregate commitment amount of $40.0 million, which may be used for revolving credit loans, swingline loans and letters of credit (the “Revolving Facility,” and together with the Term Facility, the “Credit Facility”). The Credit Facility is secured by substantially all our assets and the assets of our domestic subsidiaries.
The Term Facility proceeds were used to finance the cash portion of the consideration under the merger agreement between ANI and Novitium, repay our existing credit facility, and pay fees, costs and expenses incurred in connection with the merger. Proceeds of the Revolving Facility are expected to be used, subject to certain limitations, for working capital and other general corporate purposes.
The Term Facility matures in November 2027 and the Revolving Facility in November 2026. Each permits both base rate borrowings (“ABR Loans”) and Eurodollar rate borrowings (“Eurodollar Loans”), plus a spread of (a) 5.00% above the base rate in the case of ABR Loans under the Term Facility and 6.00% above the LIBOR Rate (as defined in the Credit Agreement, which includes a floor of 0.75%) in the case of loans under the Term Facility and (b) 3.75% above the base rate in the case of ABR Loans under the Revolving Facility and 4.75% above the LIBOR Rate (as defined in the Credit Facility) in the case of loans under the Revolving Facility. The Credit Facility has a subjective acceleration clause in case of a material adverse effect. The Term Facility includes a repayment schedule, pursuant to which $750 thousand of the loan will be paid in quarterly installments during the 12 months ended December 31, 2022. As of December 31, 2021, $3.0 million of principal of the loan was recorded as current borrowings in the consolidated balance sheet. As of December 31, 2021, we had not drawn on the Revolving Facility and $40.0 million remained available for borrowing.
Equity Financing
53
Concurrently with the execution of the Merger Agreement, on March 8, 2021, we entered into the Investment Agreement pursuant to which, on November 19, 2021, we issued and sold to the PIPE Investor, and the PIPE Investor purchased, 25,000 shares of our Series A Convertible Preferred Stock (the “PIPE Shares”), for a purchase price of $1,000 per share and an aggregate purchase price of $25 million, in a private placement issued in reliance on the exemption from registration provided by Section 4(a)(2) of the Securities Act of 1933, as amended, and/or Regulation D promulgated thereunder.
In November 2021, through a public offering, we completed the issuance and sale of 1,500,000 shares of ANI common stock, resulting in net proceeds after issuance costs of $69.7 million. The proceeds will be used to fund our Purified Cortrophin Gel commercialization efforts, including sales and marketing and consulting expenses related thereto, and for general corporate purposes.
Customer Payments
In addition to the financings in prior years, payments from customers are a significant source of cash in 2021, 2020, and 2019 and were our primary source of cash in 2021 and 2020.
Uses of Cash
Our primary cash requirements are to fund operations, including Purified Cortrophin Gel commercialization efforts, research and development programs and collaborations, to support general and administrative activities, to purchase equipment and machinery to expand our manufacturing capabilities as our product lines grow, and to expand our business and product pipeline through acquisitions of products and companies. We are continually evaluating potential asset acquisitions and business combinations. Our future capital requirements will depend on many factors, including, but not limited to:
| ● | product mix and pricing for product sales and contract manufacturing; |
| ● | pricing and payment terms with customers; |
| ● | costs of raw materials and payment terms with suppliers; |
| ● | capital expenditures and equipment purchases to support product launches; and |
| ● | business and product acquisitions. |
On November 19, 2021, we completed our previously announced acquisition of Novitium pursuant to the terms of the Merger Agreement, using $84.5 million in cash, net of $12.1 million cash acquired, 2,466,654 restricted shares of ANI common stock, and up to $46.5 million in additional contingent consideration. The contingent consideration is based on the achievement of certain milestones, including milestones on gross profit of Novitium portfolio products over a 24-month period, regulatory filings completed during this 24-month period, and a percentage of net profits on certain products that are launched in the future. As of the acquisition date, the contingent consideration had a fair value of $31.0 million.
In connection with entry into the Credit Facility, on November 19, 2021, we terminated our existing Amended and Restated Credit Agreement, dated as of December 27, 2018 (the “Prior Credit Agreement”), among the Company, as borrower, and Citizens Bank with other lenders. In connection with the termination of the Prior Credit Agreement, on November 19, 2021, we used borrowings under the Credit Facility to prepay the full amount of indebtedness under the Prior Credit Agreement, and to pay related accrued and unpaid interest, legal fees, and expenses. We made a reacquisition payment of $200.1 million, representing the remaining principal balance on the debt of $200.1 million plus certain legal fees.
In the second quarter 2021, we drew $24.0 million under the Revolver of our Prior Credit Agreement, of which $20.7 million was used to fund the acquisition of three NDAs and an ANDA and certain related inventories from Sandoz Inc. In the third quarter 2021, we utilized $8.4 million of cash on hand to settle litigation with Arbor.
54
In the first quarter of 2020, we acquired the U.S. portfolio of 23 generic products and certain commercial and development inventory and materials from Amerigen Pharmaceuticals, Ltd., for which we have used $57.4 million in cash and could make future payments of up to $25.0 million in contingent profit share payments over the next two years. The contingent payments are earned if annual gross profit exceeds a minimum threshold and are earned on a subset of the acquired products. No payment was due to Amerigen for the fiscal year ended December 31, 2020. At the time of the acquisition, the acquired portfolio included ten commercial products, three approved products with launches pending, four filed products, and four in-development products as well as a license to commercialize two approved products. The transaction was funded using cash on hand and $15.0 million of borrowings from our Revolver, of which $7.5 million was repaid in the second quarter of 2020. In the third quarter of 2020, we acquired an ANDA and certain inventories from a private company for total consideration of $4.4 million. The transaction was funded using cash on hand. In 2020, we had $6.1 million of capital expenditures.
Discussion of Cash Flows
The following table summarizes the net cash and cash equivalents provided by/(used in) operating activities, investing activities and financing activities for the periods indicated:
Year Ended December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Operating Activities | $ | 3,322 | $ | 15,267 | ||
Investing Activities | $ | (105,483) | $ | (68,322) | ||
Financing Activities | $ | 194,595 | $ | (1,439) | ||
Net Cash Provided by Operating Activities
Net cash provided by operating activities was $3.3 million for the year ended December 31, 2021, compared to $15.3 million provided by operating activities during the same period in 2020, a decrease of $11.9 million. The decrease was due to net changes in working capital and the net loss, including the incurrence of significant cash outflows related to $9.4 million of transaction expenses from the Novitium acquisition, cash outflows of $10.5 million associated with sales and marketing expenses related to Purified Cortrophin Gel launch preparation, payment for litigation settlement of $8.4 million, and payments of income taxes of $10.4 million during the year ended December 31, 2021, as compared to payments of income taxes of $5.0 million during the year ended December 31, 2020.
Net Cash Used in Investing Activities
Net cash used in investing activities for the year ended December 31, 2021 was $105.5 million, principally due to the acquisition of Novitium for $84.5 million in cash consideration, net of $12.1 million in cash acquired, the acquisition of three NDAs and an ANDA from Sandoz, Inc. for $20.7 million in consideration, and $2.6 million of capital expenditures during the period.
Net Cash Provided by / (Used In) Financing Activities
Net cash provided by financing activities was $194.6 million for the year ended December 31, 2021 compared to $1.4 million in cash used in financing activities for the year ended December 31, 2021, principally due to net proceeds of $286.5 million related to borrowings under our Credit Facility, $69.7 million related to the issuance of common shares via a public offering, $24.9 million related to the issuance of PIPE Shares, and $24.0 million in borrowings under the Revolver of our Prior Credit Agreement. These increases were tempered by the $200.1 million repayment related to all outstanding borrowings under our Prior Credit Agreement.
Contractual Obligations
We believe our available cash and cash equivalents along with our ability to generate operating cash flow and continued access to debt markets are sufficient to fund existing and planned cash requirements. Our contractual
55
obligations and commitments as of December 31, 2021 are comprised of principal payments on debt, interest payments on debt, operating leases, purchase obligations, dividends, and contingent consideration.
Our largest contractual obligation relates to our principal payments on our interest payments on our debt. As of December 31, 2021, the principal amount of our Term Facility was $300.0 million. The interest rate on our Term Facility is currently 1-month LIBOR plus 6.00%, subject to a 0.75% floor. The interest rate under the Term Facility as of December 31, 2021 is 6.75%. See Note 3, Indebtedness, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional information and timing on our principal payments on debt. We also have an interest rate swap used to manage changes in LIBOR-based interest rates underlying a portion of the borrowing under the Term Facility. Under the swap agreement, ANI pays the counterparty a fixed rate of 2.26% and receives variable 1-month LIBOR, subject to a 0.75% floor, on the outstanding notional value. As of December 31, 2021, the notional value of the interest rate swap was $168.6 million. See Note 4, Derivative Financial Instruments and Hedging Activity, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional information.
Our operating leases are for facilities and office equipment. As leases expire, we do not anticipate difficulty in negotiating renewals or finding other satisfactory space if the premise becomes unavailable. See Note 12, Commitments and Contingencies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional discussion and timing of payments related to these operating lease obligations.
Purchase obligations primarily includes contractual obligation for inventory/material purchase minimums and service agreements. We have supply agreements with three vendors that include purchase minimums. Pursuant to these agreements, we will be required to purchase a total of $12.6 million of API from these three vendors during the year ended December 31, 2022. Most of our other purchase obligations are related to purchases of information technology services, marketing arrangements or other service contracts.
Our convertible preferred stock (PIPE Shares) also accrue dividends at 6.50% per year on a cumulative basis, payable in cash or in-kind. Dividends are payable until the preferred stock is converted, either at the option of the PIPE investor, at any time, or the option of ANI, beginning two years after the November 19, 2021 issuance provided ANI’s stock price reaches a certain level. See Note 9, Mezzanine and Stockholders’ Equity, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional discussion of dividends.
Consideration of the Novitium acquisition includes $46.5 million in contingent future earn-out payments. The contingent consideration is based on the achievement of certain milestones, including milestones on gross profit of Novitium portfolio products over a 24-month period, regulatory filings completed during this 24-month period, and a percentage of net profits on certain products that are launched in the future. Payments of $25.0 million would be due if gross profit and regulatory milestones are achieved by November 30, 2023, and up to $21.5 million of payments may be made for up to ten years based on a percentage of net profits on products launched in the future. See Note 2, Business Combination, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K for additional information on our contingent consideration.
We expect to continue to incur significant expenditures in support of our commercial launch of Cortrophin, including costs related to service contracts and increased headcount.
Critical Accounting Estimates
This Management’s Discussion and Analysis of Financial Condition and Results of Operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. The SEC has defined a company’s critical accounting policies as the ones that are most important to the portrayal of the company’s financial condition and results of operations, and which require the company to make its most difficult and subjective judgments, often as a result of the
56
need to make estimates of matters that are inherently uncertain. Based on this definition, we have identified the critical accounting policies and judgments addressed below. We also have other key accounting policies, which involve the use of estimates, judgments, and assumptions that are significant to understanding our results.
Our significant accounting policies are discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K. On an ongoing basis, we evaluate these estimates and assumptions, including those described below. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results could differ from those estimates. Due to the estimation processes involved, the following summarized accounting policies and their application are considered to be critical to understanding our business operations, financial condition, and operating results.
Revenue Recognition
We recognize revenue using the following steps:
| ● | Identification of the contract, or contracts, with a customer; |
| ● | Identification of the performance obligations in the contract; |
| ● | Determination of the transaction price, including the identification and estimation of variable consideration; |
| ● | Allocation of the transaction price to the performance obligations in the contract; and |
| ● | Recognition of revenue when we satisfy a performance obligation. |
We derive our revenues primarily from sales of generic and branded pharmaceutical products. Revenue is recognized when our obligations under the terms of our contracts with customers are satisfied, which generally occurs when control of the products we sell is transferred to the customer. We estimate variable consideration after considering applicable information that is reasonably available. We generally do not have incremental costs to obtain contracts that would otherwise not have been incurred. We do not adjust revenue for the promised amount of consideration for the effects of a significant financing component because our customers generally pay us within 100 days.
Our revenue recognition accounting methodologies contain uncertainties because they require management to make assumptions and to apply judgment to estimate the amount of discounts, rebates, promotional adjustments, price adjustments, returns, chargebacks, and other potential adjustments, which are accounted for as reductions to revenue. We make these estimates based on historical experience. In addition, for our product development services revenue, we recognize revenue on a percentage of completion basis, which requires judgments related to how much work has been completed on various components our projects.
Revenue from Sales of Generic and Branded Pharmaceutical Products
Product sales consists of sales of our generic and brand pharmaceutical products. Our sole performance obligation in our contracts is to provide pharmaceutical products to customers. Our products are sold at pre-determined standalone selling prices and our performance obligation is considered to be satisfied when control of the product is transferred to the customer. Control is generally transferred to the customer upon delivery of the product to the customer, as our pharmaceutical products are generally sold on an FOB destination basis and because inventory risk and risk of ownership passes to the customer upon delivery. Payment terms for these sales are generally fewer than 100 days. We recognized $191.1 million and $195.2 million of revenue related to sales of generic and branded pharmaceutical products in 2021 and 2020, respectively.
Revenue from Distribution Agreements
From time to time, we enter into marketing and distribution agreements with third parties in which we sell products under ANDAs or NDAs owned or licensed by these third parties. These products are sold under our own label. We have assessed and determined that we control the products sold under these marketing and distribution agreements and therefore are the principal for sales under each of these marketing and distribution agreements. As a result, we recognize
57
revenue on a gross basis when control has passed to the customer and we have satisfied our performance obligation. Under these agreements, we pay these third parties a specified percentage of the gross profit earned on sales of the products. These profit-sharing percentages are recognized in cost of sales in our consolidated statements of operations and are accrued in accrued royalties in our consolidated balance sheets until payment has occurred.
Chargebacks
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, we estimate the amount of chargebacks based our actual historical experience. A number of factors influence current period chargebacks by impacting the average selling price (“ASP”) of products, including customer mix, negotiated terms, volume of off-contract purchases, and wholesale acquisition cost (“WAC”).
If actual results were not consistent with our estimates, we could be exposed to losses or gains that could be material, as changes to chargeback estimates could cause an increase or decrease in revenue recognized during the year and increase or decrease accounts receivable. If there were a 10% change in the chargeback estimates throughout the year, our net revenues would be affected by $49.2 million for the year ended December 31, 2021.
Government Rebates
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, our estimates for government rebates are based upon several factors. Our estimates for Medicaid rebates are based upon our average manufacturer price, best price, product mix, levels of inventory in the distribution channel that we expect to be subject to Medicaid rebates, and historical experience, which are invoiced in arrears by state Medicaid programs. Our estimates for Medicare rebates are based on historical experience. While such experience has allowed for reasonable estimation in the past, history may not always be an accurate indicator of future rebate experience, and trends in Medicaid and Medicare enrollment and which products are covered by Medicaid and Medicare could change.
If actual results were not consistent with our estimates, we could be exposed to losses or gains that could be material, as changes to government rebate estimates could cause an increase or decrease in revenue recognized during the year and decrease or increase the government rebate reserve. If there were a 10% change in the government rebate estimates throughout the year, our net revenues would be affected by $1.5 million for the year ended December 31, 2021.
Returns
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, our estimate for returns is based upon our historical experience with actual returns. While such experience has allowed for reasonable estimation in the past, history may not always be an accurate indicator of future returns.
If actual results were not consistent with our estimates, we could be exposed to losses or gains that could be material, as changes to returns estimates could cause an increase or decrease in revenue recognized during the year and decrease or increase the returned goods reserve. If there were a 10% change in the returns estimates throughout the year, our net revenues would be affected by $2.4 million for the year ended December 31, 2021.
Administrative Fees and Other Rebates
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, we accrue for fees and rebates by product by wholesaler, at the time of sale based on contracted rates, ASPs, and on-hand inventory counts obtained from wholesalers.
58
If actual results were not consistent with our estimates, we could be exposed to losses or gains that could be material, as changes to these estimates could cause an increase or decrease in revenue recognized during the year and increase or decrease accounts receivable. If there were a 10% change in the administrative fees estimates throughout the year, our net revenues would be affected by $3.5 million for the year ended December 31, 2021.
Prompt Payment Discounts
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, we reserve for sales discounts based on invoices outstanding, assuming, based on past experience, that 100% of available discounts will be taken.
If customers do not take 100% of available discounts as we estimate, we could need to re-adjust our methodology for calculating the prompt payment discount reserve. If there were a 10% decrease in the prompt payment discounts estimates throughout the year, our net revenues would increase by $1.6 million for the year ended December 31, 2021.
Contract Manufacturing Product Sales Revenue
Contract manufacturing arrangements consist of agreements in which we manufacture a pharmaceutical product on behalf of third party. Our performance obligation is to manufacture and provide pharmaceutical products to customers, typically pharmaceutical companies. The contract manufactured products are sold at pre-determined standalone selling prices and our performance obligations are considered to be satisfied when control of the product is transferred to the customer. Control is transferred to the customer when the product leaves our dock to be shipped to the customer, as our pharmaceutical products are sold on an FOB shipping point basis and the inventory risk and risk of ownership passes to the customer at that time. Payment terms for these sales are generally fewer than two months. We estimate returns based on historical experience. Historically, we have not had material returns for contract manufactured products. We recognized $10.0 million and $9.2 million of revenue related to sales of contract manufactured products in 2021 and 2020, respectively.
Royalties from Licensing Agreements
From time to time, we enter into transition agreements with the sellers of products we acquire, under which we license to the seller the right to sell the acquired products. Therefore, we recognize the revenue associated with sales of the underlying products as royalties. Because these royalties are sales-based, we recognize the revenue when the underlying sales occur, based on sales and gross profit information received from the sellers. Upon full transition of the products and upon launching the products under our own labels, we recognize revenue for the products as sales of generic or branded pharmaceutical products, as described above. From time to time, we enter into supply and distribution agreements with contract manufacturing customers, under which we license to the contract manufacturing customer the right to sell our products, and we are entitled to a royalty on sales made by the contract manufacturing customer under these arrangements. Therefore, we recognize the revenue associated with sales of the underlying products as royalties. Because these royalties are sales-based, we recognize the revenue when the underlying sales occur, based on sales and gross profit information received from the contract manufacturing customers.
Pursuant to a 2012 Tripartite Agreement (the “Tripartite Agreement”) between the Company, The Regents of the University of California (“The Regents”), and Cabaret Biotech Ltd., an Israeli corporation (“Cabaret”) (as assignee of Dr. Zelig Eshhar’s rights under the Tripartite Agreement), and subsequent amendments thereto and assignments thereof, we were entitled to receive a percentage of the milestone and sales royalty payments paid to Cabaret by Kite Pharma, Inc. (“Kite”), a subsidiary of Gilead Sciences, Inc., under a license agreement. Under such license agreement, Kite licensed from Dr. Eshhar and Cabaret the patent rights covered by the Tripartite Agreement and agreed to make certain payments to Cabaret based on, among other things, Kite’s sales of Yescarta®. Under the Tripartite Agreement, portions of these payments were to be distributed to The Regents and to us.
Historically, we recorded royalty income related to Yescarta® on an accrual basis utilizing our best estimate of royalties earned based upon information available in the public domain, our understanding of the various agreements governing the royalty, and other information received from time to time from the relevant parties. Generally, cash was
59
received directly from Cabaret once a year. The agreements governing this royalty were subject to multiple actions in multiple jurisdictions, including litigation between Cabaret and Kite, and separately, ANI and Cabaret. In the first quarter of 2021, we became aware that the litigation between Cabaret and Kite was dismissed. In April 2021, Cabaret and the Company settled all amounts due for amounts actually received by Cabaret or Eshhar for the licensing or use of the patent rights governed by the Kite license agreement. As a result, we recognized $11.2 million as royalties from licensing agreements in our net revenues during the three month period ended March 31, 2021. In addition, we agreed to reimburse Cabaret $0.4 million, which has been recorded as other expense, net in our consolidated statement of operations, related to certain legal expenditures incurred. We received final payment from Cabaret in May 2021. Based upon the events that led to the dismissal of the litigation between Cabaret and Kite, the Company does not expect to receive any future royalty income related to the Kite license agreement. In conjunction with payment of amounts due to us, all outstanding litigation between the Company and Cabaret were dismissed.
Product Development Services Revenue
We provide product development services to customers, which are performed over time. These services primarily relate to the technical transfer of product development to our facility in Oakville, Ontario. The duration of these technical transfer projects can be up to three years. Deposits received from these customers are recorded as deferred revenue until revenue is recognized. For contracts with no deposits and for the remainder of contracts with deposits, we invoice customers as our performance obligations are satisfied. We recognize revenue on a percentage of completion basis, which results in contract assets on our balance sheet. We recognize revenue on a proportional basis, which results in contract assets on our balance sheet. We recognized $1.3 million and $1.9 million of revenue related to product development services in 2021 and 2020, respectively.
Intangible Assets
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, our definite-lived intangible assets have a carrying value of $247.2 million as of December 31, 2021. These assets include ANDAs, NDAs and product rights, marketing and distribution rights, customer relationships, and a non-compete agreement. These intangible assets were originally recorded at fair value for business combinations and at relative fair value based on the purchase price for asset acquisitions and are stated net of accumulated amortization. As part of the Novitium acquisition on November 19, 2021, we acquired definite-lived intangible assets with a fair value of $92.3 million.
The ANDAs, NDAs and product rights, marketing and distribution rights, customer relationships, and non-compete agreement are amortized over their remaining estimated useful lives, ranging from seven to 10 years, generally based on the straight-line method unless a pattern reflecting consumption of their economic benefits is readily available. The estimated useful lives directly impact the amount of amortization expense recorded for these assets on a quarterly and annual basis.
We test for impairment of definite-lived intangible assets when events or circumstances indicate that the carrying value of the assets may not be recoverable. Judgment is used in determining when these events and circumstances arise. If we determine that the carrying value of the assets may not be recoverable, judgment and estimates are used to assess the fair value of the assets and to determine the amount of any impairment loss. If the fair value of an intangible asset is determined to be lower than its carrying value, we could be exposed to an impairment charge that could be material.
Our indefinite-lived intangible assets other than goodwill have a carrying value of $46.9 million as of December 31, 2021. These assets include in-process research and development projects (“IPR&D”) acquired in the Novitium acquisition. When an IPR&D project is completed (generally upon receipt of regulatory approval), the asset is then accounted for as a definite-lived intangible asset.
We test for impairment of indefinite-lived intangible assets at least annually, as of October 31, and whenever events or changes in circumstances indicate that the carrying amount of the asset might not be recoverable. Judgment is used in determining when these events and circumstances arise. If we determine that the carrying value of the assets may not be
60
recoverable, judgment and estimates are used to assess the fair value of the assets and to determine the amount of any impairment loss.
If the fair value of an intangible asset is determined to be lower than its carrying value, we could be exposed to an impairment charge that could be material. During the fourth quarter, 2021, we recognized a full impairment of a definite-lived ANDA asset with a remaining carrying value of $2.4 million. During the fourth quarter 2020, we recognized a full impairment of the remaining $0.4 million carrying value of a definite-lived marketing and distribution right asset.
Goodwill
As discussed in Note 1. Description of Business and Summary of Significant Accounting Policies, in the notes to the consolidated financial statements in Part II, Item 8. of this Annual Report on Form 10-K, our goodwill balance relates to our 2013 merger with BioSante Pharmaceuticals, Inc., the acquisition of WellSpring, and the acquisition of Novitium and represents the excess of the total purchase consideration over the fair value of acquired assets and assumed liabilities, using the purchase method of accounting. Goodwill is not amortized, but is subject to periodic review for impairment. As a result, the amount of goodwill is directly impacted by the estimates of the fair values of the assets acquired and liabilities assumed.
Goodwill is tested for impairment annually, as of October 31, and whenever events or changes in circumstances indicate that the carrying amount of the goodwill might not be recoverable. Judgment is used in determining when these events and circumstances arise. We perform our review of goodwill on our one reporting unit. If we determine that the carrying value of the assets may not be recoverable, judgment and estimates are used to assess the fair value of the assets and to determine the amount of any impairment loss.
The carrying value of goodwill at December 31, 2021 was $27.9 million. As part of the Novitium acquisition on November 19, 2021, we acquired goodwill of $24.3 million. We believe it is unlikely that there will be a material change in the future estimates or assumptions used to test for impairment losses on goodwill. However, if actual results are not consistent with our estimates or assumptions, we could be exposed to an impairment charge that could be material.
Contingent Consideration
The fair value of our contingent consideration was $31.0 million at December 31, 2021. The fair value of contingent consideration is remeasured to the estimated fair value each reporting period with the change recognized as an operating expense in our consolidated statements of operations. Changes in fair value can result from changes in assumptions such as discount rates, probabilities or estimates of revenue and profits, and probability of achieving regulatory milestones, as well as the passage of time. These changes resulted in a charge of $0.5 million during the year ended December 31, 2021.
Stock-Based Compensation
Our Amended and Restated 2008 Stock Incentive Plan (the “2008 Plan”) includes stock options and restricted stock, which are awarded in exchange for employee and non-employee director services. In July 2016, we commenced administration of our Employee Stock Purchase Plan (“ESPP”). We recognize the estimated fair value of stock-based awards and classify the expense where the underlying salaries are classified.
From time to time, we may grant stock options to employees through an inducement grant outside of our 2008 Plan to induce prospective employees to accept employment with us (the “Inducement Grants”). The options are granted at an exercise price equal to the fair market value of a share of our common stock on the respective grant date and are generally exercisable in four equal annual installments beginning on the first anniversary of the respective grant date. The grants are made pursuant to inducement grants outside of our stockholder approved equity plan as permitted under the Nasdaq Stock Market listing rules.
61
The following table summarizes stock-based compensation expense incurred under the 2008 Plan, Inducement Grant, and 2016 Employee Stock Purchase Plan and included in our consolidated statements of operations:
Years Ended December 31, | |||||||||
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Cost of sales | $ | 20 | $ | 137 | $ | 119 | |||
Research and development |
| 564 |
| 597 |
| 785 | |||
Selling, general, and administrative |
| 9,905 |
| 12,202 |
| 8,313 | |||
$ | 10,489 | $ | 12,936 | $ | 9,217 | ||||
Stock-based compensation cost for stock options is determined at the grant date using an option pricing model and stock-based compensation cost for restricted stock is based on the closing market price of the stock at the grant date. The value of the award is recognized as expense on a straight-line basis over the employee’s requisite service period.
Valuation of stock awards requires us to make assumptions and to apply judgment to determine the fair value of the awards. These assumptions and judgments include estimating the future volatility of our stock price and dividend yields. Changes in these assumptions can affect the fair value estimate.
Changes in estimates could affect compensation expense within individual periods. If there were to be a 10% change in our stock-based compensation expense for the year, our Loss before Benefit for Income Taxes would be affected by $1.0 million for the year ended December 31, 2021.
Income Taxes
We use the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the period that such tax rate changes are enacted.
We use a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. We have not identified any uncertain income tax positions that could have a material impact to the consolidated financial statements. We are subject to taxation in various U.S. jurisdictions, Canada, and India and remain subject to examination by taxing jurisdictions for the years 1998 and all subsequent periods due to the availability of net operating loss carryforwards. To the extent we prevail in matters for which a liability has been established, or are required to pay amounts in excess of our established liability, our effective income tax rate in a given financial statement period could be materially affected. An unfavorable tax settlement generally would require use of our cash and may result in an increase in our effective income tax rate in the period of resolution. A favorable tax settlement may reduce our effective income tax rate and would be recognized in the period of resolution.
We consider potential tax effects resulting from discontinued operations and gains and losses included in other comprehensive income and record intra-period tax allocations, when those effects are deemed material. Our effective income tax rate is also affected by changes in tax law, our level of earnings, and the results of tax audits.
Although we believe that the judgments and estimates discussed herein are reasonable, actual results could differ, and we may be exposed to losses or gains that could be material.
62
Recent Accounting Pronouncements
Recent Accounting Pronouncements Not Yet Adopted
We have evaluated all other issued and unadopted Accounting Standards Updates and believe the adoption of these standards will not have a material impact on our consolidated statements of operations, comprehensive income, balance sheets, or cash flows.
Recently Adopted Accounting Pronouncements
In August 2020, the Financial Accounting Standards Board (“FASB”) issued guidance simplifying the accounting for certain financial instruments with characteristics of liabilities and equity, including certain convertible instruments and contracts on an entity’s own equity. The new standard removes the separation models required for convertible debt with cash conversion features and convertible instruments with beneficial conversion features. It also removes certain settlement conditions that are currently required for equity contracts to qualify for the derivative scope exception and simplifies the diluted earnings per share calculation for convertible instruments. We early adopted this guidance as of January 1, 2021. The adoption of this guidance removed the requirement for an evaluation of a beneficial conversion feature related to our issuance of convertible preferred stock in November 2021 (Note 2) and will impact the calculation of diluted earnings per share in periods of net earnings.
In November 2019, the FASB issued guidance simplifying the accounting for income taxes by removing the following exceptions: 1) exception to the incremental approach for intraperiod tax allocation when there is a loss from continuing operations and income or a gain from other items, 2) exception requirement to recognize a deferred tax liability for equity method investments when a foreign subsidiary becomes an equity method investment, 3) exception to the ability not to recognize a deferred tax liability for a foreign subsidiary when a foreign equity method investment becomes a subsidiary, and 4) exception to the general methodology for calculating income taxes in an interim period when a year-to-date loss exceeds the anticipated loss for the year. The amendments also simplify accounting for income taxes by doing the following: 1) requiring that an entity recognize a franchise tax or similar tax that is partially based on income as an income-based tax and account for any incremental amount incurred as a non-income-based tax, 2) requiring that an entity evaluate when a step up in the tax basis of goodwill should be considered part of the business combination in which the book goodwill was originally recognized and when it should be considered a separate transaction, 3) specifying that an entity is not required to allocate the consolidated amount of current and deferred tax expense to a legal entity that is not subject to tax in its separate financial statements, 4) requiring that an entity reflect the effect of an enacted change in tax laws or rates in the annual effective tax rate computation in the interim period that includes the enactment date, and 5) making minor Codification improvements for income taxes related to employee stock ownership plans and investments in qualified affordable housing projects accounted for using the equity method. Most of the provisions of this guidance were to be adopted on a prospective basis. Items 2) and 3) of the “removal” provisions were to be adopted on either a full or modified retrospective basis and item 4) of the “simplifying” provisions was to be adopted on a full retrospective basis. The guidance was effective for reporting periods beginning after December 15, 2020, including interim periods within that fiscal year. We adopted this guidance as of January 1, 2021. The adoption of this guidance did not have a material impact on our consolidated financial statements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Market risks include interest rate risk, equity risk, foreign currency exchange rate risk, commodity price risk, and other relevant market rate or price risks. Of these risks, interest rate risk, equity risk, and foreign currency exchange rate risk could have a significant impact on our results of operations.
On November 19, 2021, we entered into a credit agreement (the “Credit Agreement”), which is secured by substantially all of the personal property and certain material real property owned by ANI and our wholly-owned domestic subsidiaries, and obligations under the Credit Agreement are guaranteed by certain of our wholly-owned domestic subsidiaries.
63
The Term Facility proceeds were used to finance a portion of the consideration under the Merger Agreement, repay our existing credit facility, refinance certain indebtedness of Novitium and its subsidiaries, and pay fees, costs and expenses incurred in connection with the acquisition. Proceeds of the Revolving Facility are expected to be used, subject to certain limitations, for working capital and other general corporate purposes.
The Term Facility matures on the six-year anniversary of the Closing Date and the Revolving Facility matures on the five-year anniversary of the Closing Date. The Revolving Facility and the Term Facility each permit both base rate borrowings (“ABR Loans”) and Eurodollar rate borrowings (“Eurodollar Loans”), plus a spread of (a) 5.00% above the base rate in the case of ABR Loans under the Term Facility and 6.00% above the LIBOR Rate (as defined in the Credit Facility) in the case of Eurodollar Loans under the Term Facility and (b) 3.75% above the base rate in the case of ABR Loans under the Revolving Facility and 4.75% above the LIBOR Rate (as defined in the Credit Facility) in the case of Eurodollar Loans under the Revolving Facility.
The Credit Agreement contains usual and customary representations and warranties of the parties for credit facilities of this type, subject to customary exceptions and materiality standards. In addition, we are required to maintain, a total net leverage ratio not to exceed 4.75:1.00 and, solely with respect to the Revolving Facility, (a) during the period beginning on October 1, 2022 and ending on September 30, 2023, a total net leverage ratio not to exceed 4.50:1.00 and (b) for all periods thereafter, a total net leverage ratio not to exceed 4.25:1.00.
The Credit Agreement also contains certain customary covenants and events of default, as well as, in the event of an occurrence of an event of default under the Credit Agreement, customary remedies for the lenders, including the acceleration of any amounts outstanding under the Credit Agreement.
In connection with entry into the Credit Agreement, on November 19, 2021, we terminated the Prior Credit Agreement. In connection with the termination of the Prior Credit Agreement, on the Closing Date, the Company used borrowings under the Credit Facility to prepay the full amount of indebtedness under the Prior Credit Agreement, and to pay related accrued and unpaid interest, fees and expenses. As of November 19, 2021, immediately prior to such repayment, approximately $200.0 million in aggregate principal amount was outstanding thereunder.
In April 2020, we entered into an interest rate swap with Citizens Bank, N.A. to manage our exposure to changes in LIBOR-based interest rates underlying total borrowings under term facilities related to our Prior Credit Agreement. The interest rate swap matures in December 2026. Concurrent with the termination of the Prior Credit Agreement and entry into the Credit Facility with Truist Bank, the interest rate swap with a notional value of $168.6 million was novated and is now with Truist Bank and is used to manage changes in LIBOR-based interest rates underlying a portion of the borrowing under the Term Facility. We are exposed to interest rate risk on the unhedged portion of our Term Facility and if interest rates increased or decreased by 1%, interest expense would have increased or decreased by approximately $1.3 million. If our Revolving Facility were fully drawn and interest rates increased or decreased by 1%, interest expense would have increased or decreased by approximately $0.4 million. The interest rate swap provides an effective fixed interest rate of 2.26% and has been designated as an effective cash flow hedge and therefore qualifies for hedge accounting. As a result of the interest rate swap, our exposure to interest rate volatility is minimized.
We are exposed to risks associated with changes in interest rates. The returns from certain of our cash and cash equivalents will vary as short-term interest rates change. A 100 basis-point adverse movement (decrease) in short-term interest rates would decrease the interest income earned on our cash balance in the year ended December 31, 2021 by approximately $2,000.
We are exposed to risks associated with foreign currency exchange rate risks as we remeasure certain Canadian dollar-denominated and Indian rupee-denominated transactions from ANI Pharmaceuticals Canada Inc. and our Indian subsidiary from the Canadian dollar to the U.S. dollar and the Indian-rupee to the U.S. dollar. Changes in exchange rates can positively or negatively impact our revenue, income, assets, liabilities, and equity. Currency exchange rates did not have a material impact on our revenue, income, assets, liabilities, or equity during the year ended December 31, 2021.
64
Item 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
ANI Pharmaceuticals, Inc. and Subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of ANI Pharmaceuticals, Inc. and Subsidiaries (the “Company”) as of December 31, 2021 and 2020, and the related consolidated statements of operations, comprehensive income, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2021, and the related notes (collectively referred to as the “financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the consolidated results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company’s internal control over financial reporting as of December 31, 2021 based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”), and our report dated March 15, 2022 expressed an unqualified opinion.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Evaluation of Certain Assumptions Impacting the Chargeback Accrual
As described in Note 1 to the consolidated financial statements, the Company records certain variable consideration including discounts, which are estimated at the time of sale generally using the expected value method. Amounts accrued for chargebacks as of December 31, 2021, are approximately $94.1 million and are evaluated on a quarterly basis. Management’s estimate of chargebacks is based on the inventory levels in the distribution channel as provided by wholesalers, as well as the actual average selling price for each product which is impacted by changes in customer mix, changes in negotiated terms with customers, changes in the volume of off-contract purchases, and changes in the wholesaler acquisition cost, in order to estimate the expected provision.
The principal consideration for our determination that performing procedures relating to the chargeback reserve is a critical audit matter is that there was significant judgment required by management with respect to measurement uncertainty, as the calculation of the chargeback reserve includes assumptions such as average selling price, purchasing
65
trends of distributors and historical product sales used to predict future sales. This in turn led to a high degree of auditor judgment, subjectivity and effort in applying the procedures related to those assumptions.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included assessing the design and testing the effectiveness of controls relating to the chargeback reserve, including management’s control over the assumptions used to estimate the corresponding accruals. We recalculated the chargeback accrual for a selection of products, based on a combination of Company internal data, historical actual information, and executed third-party contract. We performed a sensitivity analysis of the Company’s accrual by recalculating the accrual using our independent assumptions. We evaluated the Company’s ability to accurately estimate the accrual for chargebacks by comparing historically recorded accruals to the actual amount that was ultimately claimed by the wholesalers. We analyzed year over year trends in the reserve in comparison with revenue trends to further evaluate reasonableness of the estimate and consistency with expectations.
Accounting for Acquisition of Novitium - Valuation of Intangible Assets and Contingent Consideration
As described in Note 2 to the consolidated financial statements, the Company acquired Novitium Pharma, LLC (“Novitium”) and the transaction was accounted for using the acquisition method of accounting for business combinations. Auditing the Company’s accounting for its acquisition of Novitium was complex due to the significant estimation uncertainty required by management to determine the fair value of identified intangible assets of $139.2 million and contingent consideration of $30.5 million. The determination of the fair value of the intangible assets acquired and contingent consideration required management, with the help of a third-party valuation specialist, to make significant estimates and assumptions including the assumed net revenue growth rate, the achievement of regulatory milestones, gross profits, economic life and discount rate. The fair value of the contingent consideration represent Level 3 inputs used in measuring fair value as they are unobservable inputs with little or no available market data.
The principal consideration for our determination that the valuation of intangible assets and contingent consideration associated with the acquisition is a critical audit matter is the subjective judgment required by management in selecting the inputs and assumptions used in determining fair value. The valuation of the intangible assets and contingent consideration are subject to higher estimation uncertainty due to management’s judgment in determining key assumptions that include discount rates, probabilities of achievement of regulatory-based milestones and payments, and projected revenues and gross profits. Changes in these significant assumptions could have a significant impact on the fair value of the intangible assets and contingent consideration. This in turn led to a high degree of auditor judgment, subjectivity and effort in applying the procedures related to those assumptions.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures include assessing the design and testing the effectiveness of controls relating to the valuation report and allocation of purchase price which included management’s review of the valuation report for the completeness and mathematical accuracy of the data, and evaluating the reasonableness of assumptions used in the calculation such as economic life and discount rate. We utilized a valuation specialist to assist in evaluating the appropriateness of the Company’s valuation models developed for acquired assets and evaluating the reasonableness of significant assumptions used including the assumed net revenue growth rate, margin percentages, economic life and discount rate as compared to industry and market data. We also examined the completeness and accuracy of the underlying data supporting the significant assumptions and estimates used in the valuation report, including historical and projected financial information.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since 2013.
EISNERAMPER LLP
Philadelphia, Pennsylvania
March 15, 2022
66
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
ANI Pharmaceuticals, Inc. and Subsidiaries
Opinion on Internal Control over Financial Reporting
We have audited ANI Pharmaceuticals, Inc. and Subsidiaries’ (the “Company”) internal control over financial reporting as of December 31, 2021, based on criteria established in the Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021, based on criteria established in the Internal Control - Integrated Framework (2013) issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated balance sheets of ANI Pharmaceuticals, Inc. and Subsidiaries as of December 31, 2021 and 2020, and the related consolidated statements of operations, comprehensive income, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2021, and the related notes and our report dated March 15, 2022 expressed an unqualified opinion.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
As described in Note 2 to the consolidated financial statements, the Company acquired Novitium Pharma, LLC (“Novitium”) during the year ended December 31, 2021, and management excluded this entity from its assessment of the effectiveness of the Company’s internal control over financial reporting as of December 31, 2021 as the Company is currently in the process of integrating Novitium’s policies, processes, people, technology and operations into the consolidated company, and integrating Novitium’s operations into the consolidated internal control over financial reporting. Our audit of internal control over financial reporting of the Company also excluded an evaluation of the internal control over financial reporting of this entity.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
An entity’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. An entity’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the entity; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the entity are being made only in accordance with authorizations of management and
67
directors of the entity; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the entity’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ EisnerAmper LLP
EISNERAMPER LLP
Philadelphia, Pennsylvania
March 15, 2022
68
ANI PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Balance Sheets
(in thousands, except share and per share amounts)
| December 31, |
| December 31, | |||
2021 | 2020 | |||||
Assets | ||||||
Current Assets |
|
|
|
| ||
Cash and cash equivalents | $ | | $ | | ||
Accounts receivable, net of $ |
| |
| | ||
Inventories, net |
| |
| | ||
Prepaid income taxes |
| |
| | ||
Prepaid expenses and other current assets |
| |
| | ||
Total Current Assets |
| |
| | ||
Non-current Assets | ||||||
Property and equipment |
| |
| | ||
Accumulated depreciation | ( | ( | ||||
Property and equipment, net | | | ||||
Restricted cash |
| |
| | ||
Deferred tax assets, net of deferred tax liabilities and valuation allowance |
| |
| | ||
Intangible assets, net |
| |
| | ||
Goodwill |
| |
| | ||
Other non-current assets |
| |
| | ||
Total Assets | $ | | $ | | ||
Liabilities, Mezzanine Equity, and Stockholders’ Equity |
|
|
|
| ||
Current Liabilities |
|
|
|
| ||
Current debt, net of deferred financing costs | $ | | $ | | ||
Accounts payable |
| |
| | ||
Accrued royalties |
| |
| | ||
Accrued compensation and related expenses |
| |
| | ||
Current income taxes payable, net |
| |
| | ||
Accrued government rebates |
| |
| | ||
Returned goods reserve |
| |
| | ||
Deferred revenue |
| |
| | ||
Accrued expenses and other |
| |
| | ||
Total Current Liabilities |
| |
| | ||
Non-current Liabilities |
|
|
|
| ||
Non-current debt, net of deferred financing costs and current component |
| |
| | ||
Non-current contingent consideration |
| |
| | ||
Derivatives and other non-current liabilities |
| |
| | ||
Total Liabilities | $ | | $ | | ||
Commitments and Contingencies (Note 12) |
|
|
|
| ||
Mezzanine Equity |
|
|
|
| ||
Convertible Preferred Stock, Series A, $ |
| |
| | ||
Stockholders’ Equity |
|
|
|
| ||
Common Stock, $ |
| |
| | ||
Class C Special Stock, $ |
|
| ||||
Preferred Stock, $ |
|
| ||||
Treasury stock, |
| ( |
| ( | ||
Additional paid-in capital |
| |
| | ||
Accumulated deficit |
| ( |
| ( | ||
Accumulated other comprehensive loss, net of tax |
| ( |
| ( | ||
Total Stockholders’ Equity |
| |
| | ||
Total Liabilities, Mezzanine Equity, and Stockholders’ Equity | $ | | $ | | ||
The accompanying notes are an integral part of these consolidated financial statements.
69
ANI PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Operations
(in thousands, except per share amounts)
Years Ended December 31, | |||||||||
| 2021 |
| 2020 |
| 2019 | ||||
Net Revenues | $ | | $ | | $ | | |||
Operating Expenses |
|
|
|
|
|
| |||
Cost of sales (excluding depreciation and amortization) |
| |
| |
| | |||
Research and development |
| |
| |
| | |||
Selling, general, and administrative |
| |
| |
| | |||
Depreciation and amortization |
| |
| |
| | |||
Contingent consideration fair value adjustment | | — | — | ||||||
Legal settlement expense | | — | — | ||||||
Purified Cortrophin Gel pre-launch charges | | | | ||||||
Intangible asset impairment charge |
| |
| |
| | |||
Total Operating Expenses |
| |
| |
| | |||
Operating (Loss)/Income |
| ( |
| ( |
| | |||
Other Expense, net |
|
|
|
|
|
| |||
Interest expense, net |
| ( |
| ( |
| ( | |||
Other expense, net |
| ( |
| ( |
| ( | |||
(Loss)/Income Before Benefit for Income Taxes |
| ( |
| ( |
| | |||
Benefit for income taxes |
| |
| |
| | |||
Net (Loss)/Income | $ | ( | $ | ( | $ | | |||
Dividends on Series A Convertible Preferred Stock | $ | ( | $ | — | $ | — | |||
Net (Loss)/Income Allocated to Common Shares | $ | ( | $ | ( | $ | | |||
Basic and Diluted (Loss)/Earnings Per Share: |
|
|
|
|
|
| |||
Basic (Loss)/Earnings Per Share | $ | ( | $ | ( | $ | | |||
Diluted (Loss)/Earnings Per Share | $ | ( | $ | ( | $ | | |||
Basic Weighted-Average Shares Outstanding |
| |
| |
| | |||
Diluted Weighted-Average Shares Outstanding |
| |
| |
| | |||
The accompanying notes are an integral part of these consolidated financial statements.
70
ANI PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Comprehensive (Loss)/Income
(in thousands)
Years Ended December 31, | ||||||||||
|
| 2021 |
| 2020 |
| 2019 | ||||
Net (loss)/income | $ | ( | $ | ( | $ | | ||||
Other comprehensive income/(loss), net of tax: |
|
|
|
|
|
| ||||
Foreign currency translation adjustment | | | | |||||||
Gains/(losses) on interest rate swap, net of tax |
| |
| ( |
| ( | ||||
Total other comprehensive income/(loss), net of tax |
| |
| ( |
| ( | ||||
Total comprehensive (loss)/income, net of tax | $ | ( | $ | ( | $ | | ||||
The accompanying notes are an integral part of these consolidated financial statements.
71
ANI PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Changes in Mezzanine Equity and Stockholders’ Equity
For the Years Ended December 31, 2021, 2020, and 2019
(in thousands)
| Mezzanine Equity |
|
| Total | ||||||||||||||||||||||||||
Mezzanine Equity | Series A Convertible | Common |
| Common | Class C |
| Additional |
| Treasury |
|
| Accumulated | Mezzanine Equity | |||||||||||||||||
Series A Convertible | Preferred Stock | Stock | Stock | Special | Paid-in | Stock | Treasury | Other Comprehensive | Retained Earnings/ | and Stockholders' | ||||||||||||||||||||
Preferred Stock | Shares | Par Value | Shares | Stock | Capital | Shares | Stock | (Loss)/Gain, Net of Tax | (Accumulated Deficit) | Equity | ||||||||||||||||||||
Balance, December 31, 2018 | $ | — |
| — | $ | |
| | $ | — | $ | |
| | $ | ( | $ | ( | $ | | $ | | ||||||||
Cumulative Effect of Change in Accounting Principle, Net of Tax | — | — | — | — | — | — | — | — | — | | | |||||||||||||||||||
Stock-based Compensation Expense |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Treasury Stock Purchases for Restricted Stock Vests |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| ( |
| — |
| — |
| ( | ||||||||
Issuance of Common Shares upon Stock Option and ESPP Exercise |
| — |
| — |
| — |
| |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Issuance of Restricted Stock Awards |
| — |
| — |
| — |
| |
| — |
| ( |
| ( |
| |
| — |
| — |
| — | ||||||||
(Losses)/Gains on Interest Rate Swap | — | — | — | — | — | — | — | — | ( | — | ( | |||||||||||||||||||
Net Income | — | — | — | — | — | — | — | — | — | | | |||||||||||||||||||
Balance, December 31, 2019 | $ | — |
| — | $ | |
| | $ | — | $ | |
| | $ | ( | $ | ( | $ | | $ | | ||||||||
Cumulative Effect of Change in Accounting Principle, Net of Tax |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| ( |
| ( | ||||||||
Stock-based Compensation Expense |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Treasury Stock Purchases for Restricted Stock Vests |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| ( |
| — |
| — |
| ( | ||||||||
Issuance of Common Shares upon Stock Option and ESPP Exercise |
| — |
| — |
| — |
| |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Issuance of Restricted Stock Awards |
| — |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| — |
| — |
| — | ||||||||
Losses on Interest Rate Swap | — | — | — | — | — | — | — | — | ( | — | ( | |||||||||||||||||||
Net Loss | — | — | — | — | — | — | — | — | — | ( | ( | |||||||||||||||||||
Balance, December 31, 2020 | $ | |
| | $ | |
| | $ | — | $ | |
| | $ | ( | $ | ( | $ | ( | $ | | ||||||||
Stock-based Compensation Expense |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Treasury Stock Purchases for Restricted Stock Vests |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| ( |
| — |
| — |
| ( | ||||||||
Issuance of Common Shares upon Stock Option and ESPP Exercise |
| — |
| — |
| — |
| |
| — |
| |
| — |
| — |
| — |
| — |
| | ||||||||
Issuance of Restricted Stock Awards | — |
| — | — |
| |
| — |
| — |
| — |
| — |
| — |
| — |
| — | ||||||||||
Restricted Stock Awards Forfeitures | — | — | — | ( | — | ( | ( | — | — | — | ( | |||||||||||||||||||
Issuance of Common Stock for Novitium Acquisition | — | — | — | | — | | — | — | — | | ||||||||||||||||||||
Issuance of Common Stock in Public Offering | — | — | — | | — | | — | — | — | — | | |||||||||||||||||||
72
Dividends on Convertible Preferred Stock | — | — | — | — | — | — | — | — | — | ( | ( | |||||||||||||||||||
Issuance of Series A Convertible Preferred Stock from Mezzanine Equity | | | — | — | — | — | — | — | — | — | | |||||||||||||||||||
Other comprehensive income |
| — | — |
| — | — |
| — |
| — |
| — |
| — |
| |
| — |
| | ||||||||||
Net Loss | — | — | — | — | — | — | — | — | — | ( | ( | |||||||||||||||||||
Balance, December 31, 2021 | $ | | | $ | | | $ | — | $ | | | $ | ( | $ | ( | $ | ( | $ | |
The accompanying notes are an integral part of these consolidated financial statements.
73
ANI PHARMACEUTICALS, INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
(in thousands)
Year Ended December 31, | |||||||||
2021 | 2020 | 2019 | |||||||
Cash Flows From Operating Activities |
|
|
|
|
|
| |||
Net (loss)/income | $ | ( | $ | ( | $ | | |||
Adjustments to reconcile net loss to net cash and cash equivalents provided by operating activities: |
|
|
|
|
|
| |||
Stock-based compensation |
| |
| |
| | |||
Deferred taxes |
| ( |
| ( |
| ( | |||
Depreciation and amortization |
| |
| |
| | |||
Acquired in-process research and development ("IPR&D") |
| |
| |
| | |||
Non-cash interest |
| |
| |
| | |||
Contingent consideration fair value adjustment |
| |
| |
| | |||
Loss on extinguishment of debt | | | | ||||||
Asset impairment charge |
| |
| |
| | |||
Gain on sale of ANDAs |
| ( |
| |
| | |||
Changes in operating assets and liabilities, net of acquisitions: |
|
|
|
|
|
| |||
Accounts receivable, net |
| ( |
| ( |
| ( | |||
Inventories, net |
| |
| ( |
| ( | |||
Prepaid expenses and other current assets |
| |
| ( |
| | |||
Accounts payable |
| |
| ( |
| | |||
Accrued royalties |
| ( |
| |
| ( | |||
Current income taxes payable, net |
| ( |
| |
| ( | |||
Accrued government rebates |
| ( |
| ( |
| ( | |||
Returned goods reserve |
| |
| |
| | |||
Accrued expenses, accrued compensation, and other |
| ( |
| |
| | |||
Net Cash and Cash Equivalents Provided by Operating Activities |
| |
| |
| | |||
Cash Flows From Investing Activities |
|
|
|
|
|
| |||
Acquisition of Novitium Pharma LLC, net of cash acquired |
| ( |
| |
| | |||
Acquisition of product rights, IPR&D, and other related assets |
| ( |
| ( |
| ( | |||
Acquisition of property and equipment, net |
| ( |
| ( |
| ( | |||
Proceeds from the sale of long-lived assets |
| |
| |
| | |||
Net Cash and Cash Equivalents Used in Investing Activities |
| ( |
| ( |
| ( | |||
Cash Flows From Financing Activities |
|
|
|
|
|
| |||
Payments on Term Loan and Delayed Draw Term Loan agreements |
| ( |
| ( |
| ( | |||
Borrowings under Delayed Draw Term Loan agreement | | | | ||||||
Payments on Revolver agreement | | ( | | ||||||
Borrowings under Revolver agreement | | | | ||||||
Repayment of Prior Credit Facility | ( | | | ||||||
Borrowings under the Credit Facility | | | | ||||||
Proceeds from issuance of convertible preferred stock | | | | ||||||
Convertible preferred stock dividends paid |
| ( |
| |
| | |||
Proceeds from issuance of common stock in public offering | | | | ||||||
Cash paid for costs of share issuances | ( | | | ||||||
Proceeds from stock option exercises and ESPP purchases |
| |
| |
| | |||
Repayment of Convertible Notes |
| |
| |
| ( | |||
Payments of debt issuance costs |
| ( |
| |
| | |||
Treasury stock purchases for restricted stock vests |
| ( |
| ( |
| ( | |||
Net Cash and Cash Equivalents Provided by/(Used in) by Financing Activities |
| |
| ( |
| | |||
Net Change in Cash and Cash Equivalents |
| |
| ( |
| | |||
Cash and cash equivalents, beginning of period |
| |
| |
| | |||
Cash and cash equivalents, end of period | $ | | $ | | $ | | |||
Reconciliation of cash, cash equivalents, and restricted cash, beginning of period |
|
|
|
|
|
| |||
Cash and cash equivalents |
| |
| |
| | |||
Restricted cash |
| |
| |
| | |||
Cash, cash equivalents, and restricted cash, beginning of period |
| |
| |
| | |||
Reconciliation of cash, cash equivalents, and restricted cash, end of period |
|
|
|
|
|
| |||
Cash and cash equivalents |
| |
| |
| | |||
Restricted cash |
| |
| |
| | |||
Cash, cash equivalents, and restricted cash, end of period |
| |
| |
| | |||
Supplemental disclosure for cash flow information: |
|
|
|
|
|
| |||
Cash paid for interest, net of amounts capitalized | $ | | $ | | $ | | |||
Cash paid for income taxes | $ | | $ | | $ | | |||
Supplemental non-cash investing and financing activities: |
|
|
|
|
|
| |||
Fair value of contingent consideration in a business combination | $ | | $ | | $ | | |||
Fair value of equity issued as consideration in a business combination | $ | | $ | | $ | | |||
Acquisition of product rights, IPR&D, and other related assets included in returned goods reserve and derivatives and other non-current liabilities | $ | | $ | | $ | | |||
Property and equipment purchased and included in accounts payable | $ | | $ | | $ | | |||
The accompanying notes are an integral part of these consolidated financial statements.
74
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
1. DESCRIPTION OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Organization and Business
ANI Pharmaceuticals, Inc. and its consolidated subsidiaries, (together, “ANI,” the “Company,” “we,” “us,” or “our”) is a diversified bio-pharmaceutical company serving patients in need by developing, manufacturing, and marketing high quality branded and generic prescription pharmaceuticals, including for diseases with high unmet medical need. We are focused on delivering sustainable growth by building a successful Purified Cortrophin Gel franchise, strengthening our generics business with enhanced development capability, innovation in established brands and leveraging our North American manufacturing capabilities. Our four pharmaceutical manufacturing facilities, of which two are located in Baudette, Minnesota, one is located in East Windsor, New Jersey, and one is located in Oakville, Ontario, are together capable of producing oral solid dose products, as well as semi-solids, liquids and topicals, controlled substances, and potent products that must be manufactured in a fully-contained environment.
Basis of Presentation
The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
Principles of Consolidation
The consolidated financial statements include the accounts of ANI Pharmaceuticals, Inc. and its subsidiaries. All intercompany accounts and transactions are eliminated in consolidation.
Foreign Currency
We have subsidiaries located in Canada and India. The Canada-based subsidiary conducts its transactions in U.S. dollars and Canadian dollars, but its functional currency is the U.S. dollar. The Indian-based subsidiary generally conducts its transactions in Indian rupees, which is also its functional currency. The results of any non-U.S. dollar transactions and balances are remeasured in U.S. dollars at the applicable exchange rates during the period and resulting foreign currency transaction gains and losses are included in the determination of net income. Our gain or loss on transactions denominated in foreign currencies and the translation impact of local currencies to U.S. dollars was immaterial for the years ended December 31, 2021, 2020, and 2019. Unless otherwise noted, all references to “$” or “dollar” refer to the U.S. dollar.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. In the consolidated financial statements, estimates are used for, but not limited to, stock-based compensation, revenue recognition, allowance for credit losses, variable consideration determined based on accruals for chargebacks, administrative fees and rebates, government rebates, returns and other allowances, allowance for inventory obsolescence, valuation of financial instruments and intangible assets, accruals for contingent liabilities, including contingent consideration in acquisitions, fair value of long-lived assets, income tax provision or benefit, deferred taxes and valuation allowance, determination of right-of-use assets and lease liabilities, purchase price allocations, and the depreciable lives of long-lived assets. Because of the uncertainties inherent in such estimates, actual results may differ from those estimates. Management periodically evaluates estimates used in the preparation of the financial statements for reasonableness.
We are subject to risks and uncertainties as a result of the novel coronavirus (“COVID-19”) pandemic. We are unable to predict the impact that the COVID-19 pandemic will continue to have on our future business, financial condition, and results of operations due to numerous uncertainties. These uncertainties include the occurrence of recurring outbreaks and their severity and the duration of the pandemic, the actions taken to contain the pandemic or mitigate its impact and the direct and indirect economic effects of the pandemic and containment measures, among
75
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
others. We remain unable to predict the future impact on our estimates and assumptions. There was no material impact to these estimates or assumptions in our consolidated financial statements as of and for the years ended December 31, 2021 and 2020. Actual results could differ from those estimates, which may change our estimates in future periods. We continue to closely monitor the impact of the COVID-19 pandemic on our business.
Leases
At the inception of a contract we determine if the arrangement is, or contains, a lease. Right-of-use (“ROU”) assets represent our right to use an underlying asset for the lease term and lease liabilities represent our obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. Rent expense is recognized on a straight-line basis over the lease term.
We have made certain accounting policy elections whereby we (i) do not recognize ROU assets or lease liabilities for short-term leases (those with original terms of 12-months or less) and (ii) combine lease and non-lease elements of our operating leases. Operating lease ROU assets are included in other non-current assets and operating lease liabilities are included in accrued expenses and other and derivatives and other non-current liabilities in our consolidated balance sheets. As of December 31, 2021, we did not have any finance leases.
Comprehensive Income/(Loss)
Comprehensive (loss)/income, which is reported in the statement of comprehensive (loss)/income, consists of net (loss)/income, changes in fair value of our interest rate swap, and other comprehensive (loss)/income, net of tax.
Credit Concentration
Our customers are primarily wholesale distributors, chain drug stores, group purchasing organizations, and other pharmaceutical companies.
During the years ended December 31, 2021 and 2020 we had
The three customers represent the total percentage of net revenues as follows:
Years Ended December 31, | ||||||||||
| 2021 |
| 2020 |
| 2019 |
| ||||
Customer 1 | | % | | % | | % | ||||
Customer 2 | | % | | % | | % | ||||
Customer 3 | | % | | % | | % | ||||
Vendor Concentration
We source the raw materials for products, including active pharmaceutical ingredients (“API”), from both domestic and international suppliers. Generally, only a single source of API is qualified for use in each product due to the costs and time required to validate a second source of supply. As a result, we are dependent upon our current vendors to supply reliably the API required for on-going product manufacturing. During the year ended December 31, 2021, no single vendor represented at least 10% of inventory purchases. During the year ended December 31, 2020, we purchased approximately
76
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Revenue Recognition
We recognize revenue using the following steps:
| ● | Identification of the contract, or contracts, with a customer; |
| ● | Identification of the performance obligations in the contract; |
| ● | Determination of the transaction price, including the identification and estimation of variable consideration; |
| ● | Allocation of the transaction price to the performance obligations in the contract; and |
| ● | Recognition of revenue when we satisfy a performance obligation. |
We derive our revenues primarily from sales of generic and branded pharmaceutical products. Revenue is recognized when our obligations under the terms of our contracts with customers are satisfied, which generally occurs when control of the products we sell is transferred to the customer. We estimate variable consideration after considering applicable information that is reasonably available. We generally do not have incremental costs to obtain contracts that would otherwise not have been incurred. We do not adjust revenue for the promised amount of consideration for the effects of a significant financing component because our customers generally pay us within 100 days.
All revenue recognized in our consolidated statements of operations is considered to be revenue from contracts with customers. The following table depicts the disaggregation of revenue:
Products and Services | Years Ended December 31, | ||||||||
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Sales of generic pharmaceutical products | $ | | $ | | $ | | |||
Sales of branded pharmaceutical products |
| |
| |
| | |||
Sales of contract manufactured products |
| |
| |
| | |||
Royalties from licensing agreements |
| |
| |
| | |||
Product development services |
| |
| |
| | |||
Other |
| |
| |
| | |||
Total net revenues | $ | | $ | | $ | | |||
Timing of Revenue Recognition | Years Ended December 31, | ||||||||
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Performance obligations transferred at a point in time | $ | | $ | | $ | | |||
Performance obligations transferred over time |
| |
| |
| | |||
Total | $ | | $ | | $ | | |||
During the year ended December 31, 2021, we did not incur, and therefore did not defer, any material incremental costs to fulfill contracts. We recognized an increase of $
77
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
revenue as of December 31, 2020. For the year ended December 31, 2020, we recognized $
Revenue from Sales of Generic and Branded Pharmaceutical Products
Product sales consists of sales of our generic and brand pharmaceutical products. Our sole performance obligation in our contracts is to provide pharmaceutical products to customers. Our products are sold at pre-determined standalone selling prices and our performance obligation is considered to be satisfied when control of the product is transferred to the customer. Control is generally transferred to the customer upon delivery of the product to the customer, as our pharmaceutical products are generally sold on an FOB destination basis and because inventory risk and risk of ownership passes to the customer upon delivery. Payment terms for these sales are generally less than 100 days.
Revenue from Distribution Agreements
From time to time, we enter into marketing and distribution agreements with third parties in which we sell products under Abbreviated New Drug Applications (“ANDAs”) or New Drug Applications (“NDAs”) owned or licensed by these third parties. These products are sold under our own label. We have assessed and determined that we control the products sold under these marketing and distribution agreements and therefore are the principal for sales under each of these marketing and distribution agreements. As a result, we recognize revenue on a gross basis when control has passed to the customer and we have satisfied our performance obligation. Under these agreements, we pay these third parties a specified percentage of the gross profit earned on sales of the products. These profit-sharing percentages are recognized in cost of sales in our consolidated statements of operations and are accrued in accrued royalties in our consolidated balance sheets until payment has occurred.
Sales of our pharmaceutical products are subject to variable consideration due to chargebacks, government rebates, returns, administrative and other rebates, and cash discounts. Estimates for these elements of variable consideration require significant judgment.
Chargebacks
Chargebacks, primarily from wholesalers, result from arrangements we have with indirect customers establishing prices for products which the indirect customer purchases through a wholesaler. Alternatively, we may pre-authorize wholesalers to offer specified contract pricing to other indirect customers. Under either arrangement, we provide a chargeback credit to the wholesaler for any difference between the contracted price with the indirect customer and the wholesaler’s invoice price, typically Wholesale Acquisition Cost (“WAC”).
Chargeback credits are calculated as follows:
Prior period chargebacks claimed by wholesalers are analyzed to determine the actual average selling price ("ASP") for each product. This calculation is performed by product by wholesaler. ASPs can be affected by several factors such as:
| ● | A change in customer mix |
| ● | A change in negotiated terms with customers |
| ● | A change in the volume of off-contract purchases |
| ● | Changes in WAC |
As necessary, we adjust ASPs based on anticipated changes in the factors above.
The difference between ASP and WAC is recorded as a reduction in both gross revenues in our consolidated statements of operations and accounts receivable in the consolidated balance sheets, at the time we recognize revenue from the product sale.
To evaluate the adequacy of our chargeback accruals, we obtain on-hand inventory counts from the wholesalers. This inventory is multiplied by the chargeback amount, the difference between ASP and WAC, to arrive at total
78
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
expected future chargebacks, which is then compared to the chargeback accruals. We continually monitor chargeback activity and adjust ASPs when we believe that actual selling prices will differ from current ASPs.
Government Rebates
Our government rebates reserve consists of estimated payments due to governmental agencies for purchases made by third parties under various governmental programs. The two largest government programs that impact our net revenue and our government rebates reserve are federal and state Medicaid rebate programs and Medicare.
We participate in certain qualifying federal and state Medicaid rebate programs whereby discounts and rebates are provided to participating programs after the final dispensing of the product by a pharmacy to a Medicaid plan participant. Medicaid rebates are typically billed up to 120 days after the product is shipped. Medicaid rebate amounts per product unit are established by law, based on the Average Manufacturer Price (“AMP”), which is reported on a monthly and quarterly basis, and, in the case of branded products, best price, which is reported on a quarterly basis. Our Medicaid reserves are based on expected claims from state Medicaid programs. Estimates for expected claims are driven by patient usage, sales mix, calculated AMP or best price, as well as inventory in the distribution channel that will be subject to a Medicaid rebate. As a result of the delay between selling the products and rebate billing, our Medicaid rebate reserve includes both an estimate of outstanding claims for end-customer sales that have occurred but for which the related claim has not been billed, as well as an estimate for future claims that will be made when inventory in the distribution channel is sold through to plan participants.
Many of our products are also covered under Medicare. We, like all pharmaceutical companies, must provide a discount for any products sold under NDAs to Medicare Part D participants. This applies to all products sold under NDAs, regardless of whether the products are marketed as branded or generic. Our estimates for these discounts are based on historical experience with Medicare rebates for our products. While such experience has allowed for reasonable estimations in the past, history may not always be an accurate indicator of future rebates. Medicare rebates are typically billed up to 120 days after the product is shipped. As a result of the delay between selling the products and rebate billing, our Medicare rebate reserve includes both an estimate of outstanding claims for end-customer sales that have occurred but for which the related claim has not been billed, as well as an estimate for future claims that will be made when inventory in the distribution channel is sold through to Medicare Part D participants.
To evaluate the adequacy of our government rebate reserves, we review the reserves on a quarterly basis against actual claims data to ensure the liability is fairly stated. We continually monitor our government rebate reserve and adjust our estimates if we believe that actual government rebates may differ from our established accruals. Accruals for government rebates are recorded as a reduction to gross revenues in our consolidated statements of operations and as an increase to accrued government rebates in the consolidated balance sheets.
Returns
We maintain a return policy that allows customers to return product within a specified period prior to and subsequent to the expiration date. Generally, product may be returned for a period beginning six months prior to its expiration date to up to one year after its expiration date. Our product returns are settled through the issuance of a credit to the customer. Our estimate for returns is based upon historical experience with actual returns. While such experience has allowed for reasonable estimation in the past, history may not always be an accurate indicator of future returns. We continually monitor our estimates for returns and make adjustments when we believe that actual product returns may differ from the established accruals. Accruals for returns are recorded as a reduction to gross revenues in our consolidated statements of operations and as an increase to the return goods reserve in the consolidated balance sheets.
Administrative Fees and Other Rebates
Administrative fees or rebates are offered to wholesalers, group purchasing organizations, and indirect customers. We accrue for fees and rebates, by product by wholesaler, at the time of sale based on contracted rates and ASPs.
79
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
To evaluate the adequacy of our administrative fee accruals, we obtain on-hand inventory counts from the wholesalers. This inventory is multiplied by the ASPs to arrive at total expected future sales, which is then multiplied by contracted rates. The result is then compared to the administrative fee accruals. We continually monitor administrative fee activity and adjust our accruals when we believe that actual administrative fees will differ from the accruals. Accruals for administrative fees and other rebates are recorded as a reduction in both gross revenues in our consolidated statements of operations and accounts receivable in the consolidated balance sheets.
Prompt Payment Discounts
We often grant sales discounts for prompt payment. The reserve for prompt payment discounts is based on invoices outstanding. We assume, based on past experience, that all available discounts will be taken. Accruals for prompt payment discounts are recorded as a reduction in both gross revenues in our consolidated statements of operations and accounts receivable in the consolidated balance sheets.
The following table summarizes activity in the consolidated balance sheets for accruals and allowances for the years ended December 31, 2021, 2020, and 2019:
Accruals for Chargebacks, Returns, and Other Allowances | |||||||||||||||
Administrative | Prompt | ||||||||||||||
Government | Fees and Other | Payment | |||||||||||||
(in thousands) |
| Chargebacks |
| Rebates |
| Returns |
| Rebates |
| Discounts | |||||
Balance at December 31, 2019 (1) | $ | | $ | | $ | | $ | | $ | | |||||
Accruals/Adjustments |
| |
| |
| |
| |
| | |||||
Credits Taken Against Reserve |
| ( | ( |
| ( |
| ( |
| ( | ||||||
Balance at December 31, 2020 (1) | $ | | $ | | $ | | $ | | $ | | |||||
Accruals/Adjustments |
| |
| |
| |
| |
| | |||||
Credits Taken Against Reserve |
| ( | ( |
| ( |
| ( |
| ( | ||||||
Balance at December 31, 2021 (1) | $ | | $ | | $ | | $ | | $ | | |||||
Contract Manufacturing Product Sales Revenue
Contract manufacturing arrangements consist of agreements in which we manufacture a pharmaceutical product on behalf of a third party. Our performance obligation is to manufacture and provide pharmaceutical products to customers, typically pharmaceutical companies. The contract manufactured products are sold at pre-determined standalone selling prices and our performance obligations are considered to be satisfied when control of the product is transferred to the customer. Control is transferred to the customer when the product leaves our dock to be shipped to the customer, as our contract manufactured pharmaceutical products are sold on an FOB shipping point basis and the inventory risk and risk of ownership passes to the customer at that time. Payment terms for these sales are generally fewer than two months. We estimate returns based on historical experience. Historically, we have not had material returns for contract manufactured products.
As of December 31, 2021, the aggregate amount of the transaction price allocated to the remaining performance obligations for all open contract manufacturing customer contracts was $
80
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Royalties from Licensing Agreements
From time to time, we enter into transition agreements with the sellers of products we acquire, under which we license to the seller the right to sell the acquired products. Therefore, we recognize the revenue associated with sales of the underlying products as royalties. Because these royalties are sales-based, we recognize the revenue when the underlying sales occur, based on sales and gross profit information received from the sellers. Upon full transition of the products and upon launching the products under our own labels, we recognize revenue for the products as sales of generic or branded pharmaceutical products, as described above. From time to time, we enter into supply and distribution agreements with contract manufacturing customers, under which we license to the contract manufacturing customer the right to sell our products, and we are entitled to a royalty on sales made by the contract manufacturing customer under these arrangements. Therefore, we recognize the revenue associated with sales of the underlying products as royalties. Because these royalties are sales-based, we recognize the revenue when the underlying sales occur, based on sales and gross profit information received from the contract manufacturing customers.
Pursuant to a 2012 Tripartite Agreement (the “Tripartite Agreement”) between the Company, The Regents of the University of California (“The Regents”), and Cabaret Biotech Ltd., an Israeli corporation (“Cabaret”) (as assignee of Dr. Zelig Eshhar’s rights under the Tripartite Agreement), and subsequent amendments thereto and assignments thereof, we were entitled to receive a percentage of the milestone and sales royalty payments paid to Cabaret by Kite Pharma, Inc. (“Kite”), a subsidiary of Gilead Sciences, Inc., under a license agreement. Under such license agreement, Kite licensed from Dr. Eshhar and Cabaret the patent rights covered by the Tripartite Agreement and agreed to make certain payments to Cabaret based on, among other things, Kite’s sales of Yescarta®. Under the Tripartite Agreement, portions of these payments were to be distributed to The Regents and to us.
Historically, we recorded royalty income related to Yescarta® on an accrual basis utilizing our best estimate of royalties earned based upon information available in the public domain, our understanding of the various agreements governing the royalty, and other information received from time to time from the relevant parties. Generally, cash was received directly from Cabaret once a year. The agreements governing this royalty were subject to multiple actions in multiple jurisdictions, including litigation between Cabaret and Kite, and separately, ANI and Cabaret. In the first quarter of 2021, we became aware that the litigation between Cabaret and Kite was dismissed. In April 2021, Cabaret and the Company settled all amounts due for amounts actually received by Cabaret or Eshhar for the licensing or use of the patent rights governed by the Kite license agreement. As a result, we recognized $
Product Development Services Revenue
We provide product development services to customers, which are performed over time. These services primarily relate to the technical transfer of product development to our facility in Oakville, Ontario. The duration of these technical transfer projects can be up to three years. Deposits received from these customers are recorded as deferred revenue until revenue is recognized. For contracts with no deposits and for the remainder of contracts with deposits, we invoice customers as our performance obligations are satisfied. We recognize revenue on a percentage of completion basis, which results in contract assets on our balance sheet. As of December 31, 2021, the aggregate amount of the transaction price allocated to the remaining performance obligations for all product development services contracts was less than $
Cash, Cash Equivalents, and Restricted Cash
We consider all highly liquid instruments with maturities of three months or less when purchased to be cash equivalents. All interest bearing and non-interest bearing accounts are guaranteed by the Federal Deposit Insurance
81
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Corporation (“FDIC”) up to $
In April 2016, we purchased the rights, title, and interest in the NDA for Inderal LA, as well as certain documentation, trademark rights, and finished goods from Cranford Pharmaceuticals, LLC for $
Accounts Receivable
We extend credit to customers on an unsecured basis. We measure expected credit losses on our financial assets at amortized cost, including trade and unbilled receivables, on a collective basis, based on their similar risk characteristics. Expected credits losses are based on historical credit loss experience, review of the current aging or status of accounts receivable and current and forward-looking views from an economic and industry perspective. We determine trade receivables to be delinquent when greater than 30 days past due. Receivables are written off when it is determined that amounts are uncollectible. Our allowance for credit losses was immaterial as of December 31, 2021 and 2020.
Inventories
Inventories consist of raw materials, packaging materials, work-in-progress, and finished goods. Inventories are stated at the lower of standard cost or net realizable value. We periodically review and adjust standard costs, which generally approximate weighted average cost.
Property and Equipment
Property and equipment are recorded at cost. Expenditures for repairs and maintenance are charged to expense as incurred. Depreciation is recorded on a straight-line basis over estimated useful lives as follows:
Buildings and improvements |
| - |
| years | ||
Machinery, furniture, and equipment | years |
Construction in progress consists of multiple projects, primarily related to new equipment to expand our manufacturing capability as our product lines grow. Construction in progress includes the cost of construction and other direct costs attributable to the construction, along with capitalized interest. Depreciation is not recorded on construction in progress until such time as the assets are placed in service.
We review property and equipment for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of the long-lived asset is measured by a comparison of the carrying amount of the asset to future undiscounted net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the estimated fair value of the assets.
Intangible Assets
Definite-lived intangible assets consist of acquired ANDAs for previously commercialized and marketed drug products, acquired approved ANDAs for generic products yet to be commercialized, an acquired development package for a generic drug product, a license, supply and distribution agreement for a generic drug product, acquired product rights for generic products, acquired NDAs and product rights for branded products, acquired marketing and distribution rights, acquired customer relationships, and a non-compete agreement. They are stated at cost, net of amortization, generally using the straight-line method over the expected useful lives of the intangible assets.
82
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
The definite-lived ANDAs, NDAs and product rights, marketing and distribution rights, customer relationships, and non-compete agreement are stated at cost, net of amortization, and generally amortized over their remaining estimated useful lives, ranging from to
Our indefinite-lived intangible assets other than goodwill include in-process research and development (“IPR&D”) projects. IPR&D intangible assets represent the fair value of technology acquired in a business combination for which the technology projects are incomplete but have substance. IPR&D acquired in a business combination is initially capitalized as an indefinite-lived intangible asset until the project is complete, which is generally when we receive regulatory approval for a product. Upon approval, we determine the useful life of the asset and begin amortizing the value over that life. IPR&D acquired in a purchase of assets rather than a business is expensed as incurred. We test for impairment of indefinite-lived intangible assets at least annually, as of October 31, and whenever events or changes in circumstances indicate that the carrying amount of the asset might not be recoverable. Judgment is used in determining when these events and circumstances arise. If we determine that the carrying value of the assets may not be recoverable, judgment and estimates are used to assess the fair value of the assets and to determine the amount of any impairment loss. No events or circumstances arose in 2021 that indicated that the carrying value of any of our other indefinite-lived intangible assets may not be recoverable.
Goodwill
Goodwill relates to the 2013 merger with BioSante Pharmaceuticals, Inc. and the acquisitions of WellSpring and Novitium, and represents the excess of the total purchase consideration over the fair value of acquired assets and assumed liabilities, using the purchase method of accounting. Goodwill is not amortized, but is subject to periodic review for impairment. Goodwill is reviewed for impairment annually, as of October 31, and whenever events or changes in circumstances indicate that the carrying amount of the goodwill might not be recoverable. We perform our review of goodwill on our one reporting unit.
Before employing detailed impairment testing methodologies, we first evaluate the likelihood of impairment by considering qualitative factors relevant to our reporting unit. When performing the qualitative assessment, we evaluate events and circumstances that would affect the significant inputs used to determine the fair value of the goodwill. Events and circumstances evaluated include macroeconomic conditions that could affect us, industry and market considerations for the generic pharmaceutical industry that could affect us, cost factors that could affect our performance, our financial performance (including share price), and consideration of any company-specific events that could negatively affect us, our business, or the fair value of our business. If we determine that it is more likely than not that goodwill is impaired, we will then apply detailed testing methodologies. Otherwise, we will conclude that no impairment has occurred.
Detailed impairment testing involves comparing the fair value of our one reporting unit to its carrying value, including goodwill. Fair value reflects the price a market participant would be willing to pay in a potential sale of ANI. If the fair value exceeds carrying value, then it is concluded that no goodwill impairment has occurred. If the carrying value of the reporting unit were to exceed its fair value, we would recognize an impairment charge for the amount by which the carrying amount exceeded the reporting unit’s fair value. The loss recognized would not exceed the total amount of goodwill allocated to that reporting unit.
83
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Collaborative Arrangements
At times, we have entered into arrangements with various commercial partners to further business opportunities. In collaborative arrangements such as these, when we are actively involved and exposed to the risks and rewards of the activities and are determined to be the principal participant in the collaboration, we classify third party costs incurred and revenues in our consolidated statements of operations on a gross basis. Otherwise, third party revenues and costs generated by collaborative arrangements are presented on a net basis. Payments between us and the other participants are recorded and classified based on the nature of the payments.
Royalties
We have entered profit-sharing arrangements with third parties in which we sell products under ANDAs or NDAs owned or licensed by these third parties. Under these agreements, we pay these third parties a specified percentage of the gross profit earned on sales of the products. These profit-sharing percentages are recorded in cost of sales in our consolidated statements of operations when the associated revenue is recognized and are recorded in accrued royalties in our consolidated balance sheets when the associated revenue is recognized and until payment has occurred.
Research and Development Expenses
Research and development costs are expensed as incurred and primarily consist of expenses relating to product development. Research and development costs totaled $
Stock-Based Compensation
We have a stock-based compensation plan that includes stock options and restricted stock, which are awarded in exchange for employee and non-employee director services. From time to time, we may make awards through an inducement grant outside of our plan to induce prospective employees to accept employment with us. These grants are made pursuant to inducement grants outside of our shareholder approved equity plan as permitted under the Nasdaq Stock Market listing rules. Stock-based compensation cost for stock options is determined at the grant date using an option pricing model and stock-based compensation cost for restricted stock is based on the closing market price of the stock at the grant date. The value of the award is recognized as expense on a straight-line basis over the employee’s requisite service period and classified where the underlying salaries are classified. We also account for forfeitures as they occur. We recognize excess tax benefits or tax deficiencies as a component of our current period provision for income taxes.
In addition, in July 2016, we commenced administration of our Employee Stock Purchase Plan (“ESPP”). We recognize the estimated fair value of stock-based compensation awards and classify the expense where the underlying salaries are classified.
We incurred $
Valuation of stock awards requires us to make assumptions and to apply judgment to determine the fair value of the awards. These assumptions and judgments include estimating the future volatility of our stock price and dividend yields. Changes in these assumptions can affect the fair value estimate.
Income Taxes
We use the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that are expected to be in effect when the differences are expected to
84
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
reverse. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in the period that such tax rate changes are enacted.
The measurement of a deferred tax asset is reduced, if necessary, by a valuation allowance if it is more likely than not that some portion or all of the deferred tax asset will not be realized. We have provided a valuation allowance against certain of our state net operating loss (“NOL”) carryforwards that are not expected to be used during the carryforward periods. As of December 31, 2018, we had also provided a valuation allowance against ANI Canada’s net deferred tax assets of $
We have not provided for deferred taxes related to any difference between the tax basis in the shares of ANI Canada and the financial reporting basis in those shares since it has the intent and ability to indefinitely reinvest ANI Canada’s earnings and not repatriate those earnings.
We use a recognition threshold and a measurement attribute for the financial statement recognition and measurement of tax positions taken or expected to be taken in a tax return. For those benefits to be recognized, a tax position must be more-likely-than-not to be sustained upon examination by taxing authorities. We have not identified any uncertain income tax positions that could have a material impact on the consolidated financial statements.
We recognize interest and penalties accrued on any unrecognized tax exposures as a component of income tax expense; we did not have any material amounts accrued as of December 31, 2021, 2020, and 2019. We are subject to taxation in various U.S. jurisdictions, Canada, and India, and all of our income tax returns remain subject to examination by tax authorities due to the availability of NOL carryforwards.
We consider potential tax effects resulting from discontinued operations and for gains and losses in other comprehensive income and record intra-period tax allocations, when those effects are deemed material. We previously entered into an interest rate swap agreement (Note 4) that we have designated as a cash flow hedge designed to manage exposure to changes in LIBOR-based interest rate underlying our variable rate debt. Due to the effective nature of the hedge, the initial fair value of the hedge and subsequent changes in the fair value of the hedge are recognized in accumulated other comprehensive loss, net of tax in the consolidated balance sheets. Income taxes are allocated to the hedge component of accumulated other comprehensive income based on appropriate intra-period tax allocations when those effects are deemed material.
Earnings (Loss) per Share
Basic earnings (loss) per share is computed by dividing net income (loss) available to common shareholders by the weighted-average number of shares of common stock outstanding during the period.
For periods of net income, and when the effects are not anti-dilutive, we calculate diluted earnings (loss) per share by dividing net income available to common shareholders by the weighted-average number of shares outstanding plus the impact of all potential dilutive common shares, consisting primarily of common stock options, shares to be purchased under our ESPP, unvested restricted stock awards under the treasury stock method, and convertible preferred stock using the if-converted method. For periods of net loss, diluted loss per share is calculated similarly to basic loss per share.
Our unvested restricted shares and convertible preferred stock shares contain non-forfeitable rights to dividends, and therefore are considered to be participating securities; in periods of net income, the calculation of basic and diluted earnings (loss) per share excludes from the numerator net income (but not net loss) attributable to the unvested restricted shares and the common shares assumed converted from the preferred shares and excludes the impact of those shares from the denominator.
85
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
For purposes of determining diluted earnings (loss) per share in 2019, we elected a policy to settle the principal portion of our
Earnings per share for the years ended December 31, 2021, 2020, and 2019 are calculated for basic and diluted earnings (loss) per share as follows:
Basic | Diluted | |||||||||||||||||
(in thousands, except per share amounts) | Years Ended December 31, | Years Ended December 31, | ||||||||||||||||
| 2021 |
| 2020 |
| 2019 |
| 2021 |
| 2020 |
| 2019 | |||||||
Net (loss)/income | $ | ( | $ | ( | $ | | $ | ( | $ | ( | $ | | ||||||
Net income allocated to participating securities |
| — |
| — |
| ( |
| — |
| — |
| ( | ||||||
Dividends on Series A convertible preferred stock | ( | — | — | ( | — | — | ||||||||||||
Net (loss)/income allocated to common shares | $ | ( | $ | ( | $ | | $ | ( | $ | ( | $ | | ||||||
Basic Weighted-Average Shares Outstanding |
| |
| |
| |
| |
| |
| | ||||||
Dilutive effect of stock options and ESPP |
| — |
| — |
| | ||||||||||||
Dilutive effect of Notes | — | — | | |||||||||||||||
Diluted Weighted-Average Shares Outstanding |
| |
| |
| | ||||||||||||
(Loss)/Income per share | $ | ( | $ | ( | $ | | $ | ( | $ | ( | $ | | ||||||
The number of anti-dilutive shares, which have been excluded from the computation of diluted earnings (loss) per share, were
Hedge Accounting
At times we use derivative financial instruments to hedge our exposure to interest rate risks. All derivative financial instruments are recognized as either assets or liabilities at fair value on the consolidated balance sheet and are classified as current or non-current based on the scheduled maturity of the instrument.
When we enter into a hedge arrangement and intend to apply hedge accounting, we formally document the hedge relationship and designate the instrument for financial reporting purposes as a fair value hedge, a cash flow hedge, or a net investment hedge. When we determine that a derivative financial instrument qualifies as a cash flow hedge and is effective, the changes in fair value of the instrument are recorded in accumulated other comprehensive (loss)/income, net of tax in our consolidated balance sheets and will be reclassified to earnings when the hedged item affects earnings.
Contingent Consideration
86
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
The terms of the acquisition agreement between ANI and Novitium Pharma LLC include the potential payment of future consideration that is contingent upon the achievement of certain regulatory and financial performance milestones. At acquisition date, we recorded this contingent consideration at fair value based on the additional consideration expected to be transferred, which is based on the estimate of probability-weighted future cash flows as discounted to present value. Significant inputs used in the measurement of the fair value include discount rates, probabilities of achievement of regulatory-based milestones and payments, and projected revenues and gross profits. The discount rates are derived using accepted valuation methodologies. The probability of achievement of regulatory milestones is based on historical and projected success rates. The projected revenues and gross profits are based on our internal forecasts and long-term plans. We remeasure the fair value of the contingent consideration each reporting period using Level 3 inputs, as discussed further below. Changes in fair value, which incorporate changes in assumptions and the passage of time, are recognized as an operating expense in our consolidated statement of operations. As payments are not expected to be made shortly after the acquisition, any future payment of contingent consideration will be reported as a financing cash flow for amounts paid up to the acquisition-date fair value of the consideration, and as an operating cash outflow for any amounts in excess of the acquisition-date fair value in our consolidated statement of cash flows.
Fair Value of Financial Instruments
Our consolidated balance sheets include various financial instruments (primarily cash and cash equivalents, prepaid expenses, accounts receivable, accounts payable, accrued expenses, and other current liabilities) that are carried at cost and that approximate fair value. Fair value is the price that would be received from the sale of an asset or paid to transfer a liability assuming an orderly transaction in the most advantageous market at the measurement date. U.S. GAAP establishes a hierarchical disclosure framework which prioritizes and ranks the level of observability of inputs used in measuring fair value. These tiers include:
| ● | Level 1—Quoted prices (unadjusted) in active markets that are accessible at the measurement date for identical assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| ● | Level 2—Observable market-based inputs other than quoted prices in active markets for identical assets or liabilities. |
| ● | Level 3—Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
See Note 8 for additional information regarding fair value.
Geographic Information
Based on the distinct nature of our operations, our internal management structure, and the financial information that is evaluated regularly by our Chief Operating Decision Maker, we determined that we operate in
The following table depicts our revenue by geographic operations during the following periods:
(in thousands) | Years Ended December 31, | ||||||||
Location of Operations |
| 2021 |
| 2020 |
| 2019 | |||
United States | $ | | $ | | $ | | |||
Canada |
| |
| |
| | |||
Total Revenue | $ | | $ | | $ | | |||
The following table depicts our property and equipment, net according to geographic location as of:
(in thousands) | December 31, 2021 | December 31, 2020 | ||||
United States | $ | | $ | | ||
87
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Canada |
| |
| | ||
India |
| |
| — | ||
Total property and equipment, net | $ | | $ | |
Recent Accounting Pronouncements
Recent Accounting Pronouncements Not Yet Adopted
We have evaluated all other issued and unadopted Accounting Standards Updates and believe the adoption of these standards will not have a material impact on our consolidated statements of operations, comprehensive income, balance sheets, or cash flows.
Recently Adopted Accounting Pronouncements
In August 2020, the Financial Accounting Standards Board (“FASB”) issued guidance simplifying the accounting for certain financial instruments with characteristics of liabilities and equity, including certain convertible instruments and contracts on an entity’s own equity. The new standard removes the separation models required for convertible debt with cash conversion features and convertible instruments with beneficial conversion features. It also removes certain settlement conditions that are currently required for equity contracts to qualify for the derivative scope exception and simplifies the diluted earnings per share calculation for convertible instruments. We early adopted this guidance as of January 1, 2021. The adoption of this guidance removed the requirement for an evaluation of a beneficial conversion feature related to our issuance of convertible preferred stock in November 2021 and will impact the calculation of diluted earnings per share in periods of net earnings.
In November 2019, the FASB issued guidance simplifying the accounting for income taxes by removing the following exceptions: 1) exception to the incremental approach for intraperiod tax allocation when there is a loss from continuing operations and income or a gain from other items, 2) exception requirement to recognize a deferred tax liability for equity method investments when a foreign subsidiary becomes an equity method investment, 3) exception to the ability not to recognize a deferred tax liability for a foreign subsidiary when a foreign equity method investment becomes a subsidiary, and 4) exception to the general methodology for calculating income taxes in an interim period when a year-to-date loss exceeds the anticipated loss for the year. The amendments also simplify accounting for income taxes by doing the following: 1) requiring that an entity recognize a franchise tax or similar tax that is partially based on income as an income-based tax and account for any incremental amount incurred as a non-income-based tax, 2) requiring that an entity evaluate when a step up in the tax basis of goodwill should be considered part of the business combination in which the book goodwill was originally recognized and when it should be considered a separate transaction, 3) specifying that an entity is not required to allocate the consolidated amount of current and deferred tax expense to a legal entity that is not subject to tax in its separate financial statements, 4) requiring that an entity reflect the effect of an enacted change in tax laws or rates in the annual effective tax rate computation in the interim period that includes the enactment date, and 5) making minor Codification improvements for income taxes related to employee stock ownership plans and investments in qualified affordable housing projects accounted for using the equity method. Most of the provisions of this guidance were to be adopted on a prospective basis. Items 2) and 3) of the “removal” provisions were to be adopted on either a full or modified retrospective basis and item 4) of the “simplifying” provisions was to be adopted on a full retrospective basis. The guidance was effective for reporting periods beginning after December 15, 2020, including interim periods within that fiscal year. We adopted this guidance as of January 1, 2021. The adoption of this guidance did not have a material impact on our consolidated financial statements.
2. BUSINESS COMBINATION
Summary
On November 19, 2021, we completed our previously announced acquisition of all of the interests of Novitium pursuant to the terms of the Agreement and Plan of Merger, dated as of March 8, 2021, for cash consideration,
88
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
on the date of closing and discounted for lack of marketability due to restrictions on shares, and up to $
Purchase consideration consisted of the following:
| (in thousands) | ||
Cash consideration | $ | | |
Repayment of Novitium debts |
| | |
Fair value of restricted shares |
| | |
Fair value of contingent consideration |
| | |
Gross consideration | $ | | |
Cash acquired | | ||
Net consideration | $ | | |
The cash consideration was funded in part by borrowings under our new credit facility (Note 3) and through issuance of PIPE convertible preferred stock shares (Note 9). We acquired Novitium due to its proven track record of being a research and development growth engine capable of fueling sustainable growth, to expand our research and development pipeline via niche opportunities, to enhance our contract development and manufacturing organization (“CDMO”) business and U.S. based manufacturing capacity, and to diversify our revenue base.
The preliminary allocation of the fair value of the Novitium acquisition is shown in the table below. The allocation of the fair value will be finalized when the valuation is completed and the differences will be trued up for the final allocated amounts.
| (in thousands) | ||
Total Purchase Consideration | $ | | |
Cash and cash equivalents |
| | |
Accounts receivable |
| | |
Inventories |
| | |
Prepaid expenses and other current assets |
| | |
Property and equipment |
| | |
Intangible assets | | ||
Goodwill |
| | |
Other non-current assets | | ||
Total assets acquired |
| | |
Accounts payable |
| | |
Accrued expense and other current liabilities | | ||
Accrued compensation and other related expenses | | ||
Accrued government rebates | | ||
Returned goods reserve | | ||
Other non-current liabilities | | ||
Total liabilities assumed |
| | |
Net assets acquired | $ | | |
The net assets were recorded at their estimated fair value. In valuing acquired assets and liabilities, fair value estimates were based primarily on future expected cash flows, market rate assumptions for contractual obligations,
89
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
and appropriate discount rates. In connection with the acquisition, we recognized $
Goodwill is considered an indefinite-lived asset and relates primarily to intangible assets that do not qualify for separate recognition, such as the assembled workforce and synergies between the entities. Goodwill established as a result of the acquisition is tax deductible in the U.S.
Novitium operations generated $
Pro Forma Consolidated Financial Information (unaudited)
The following unaudited pro forma consolidated financial information summarizes the results of operations for the periods indicated as if the Novitium acquisition had been completed as of January 1, 2020.
Years Ended December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Net revenues | $ | | $ | | ||
Net loss | $ | ( | $ | ( | ||
Transaction Costs
In conjunction with the acquisition, we incurred approximately $
Restricted Shares
The Novitium acquisition consideration included
3. INDEBTEDNESS
Credit Facility
On November 19, 2021, the Company, as borrower, entered into a credit agreement (the “Credit Agreement”) with Truist Bank and other lenders, which provides for credit facilities consisting of (i) a senior secured term loan facility in an aggregate principal amount of $
The Term Facility proceeds were used to finance the cash portion of the consideration under the merger agreement between ANI and Novitium, repay our existing credit facility, and pay fees, costs and expenses incurred in connection with the merger. Proceeds of the Revolving Facility are expected to be used, subject to certain limitations, for working capital and other general corporate purposes.
The Term Facility matures in November 2027 and the Revolving Facility in November 2026. Each permits both base rate borrowings (“ABR Loans”) and Eurodollar rate borrowings (“Eurodollar Loans”), plus a spread of (a)
90
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
defined in the Credit Agreement) in the case of LIBOR loans under the Term Facility and (b)
We incurred $
In connection with entry into the Credit Facility, on November 19, 2021, we terminated our existing Amended and Restated Credit Agreement, dated as of December 27, 2018 (the “Prior Credit Agreement”), among the Company, as borrower, and Citizens Bank with other lenders. In connection with the termination of the Prior Credit Agreement, on November 19, 2021, we used borrowings under the Credit Facility to prepay the full amount of indebtedness under the Prior Credit Agreement, and to pay related accrued and unpaid interest, fees, and expenses. The repayment and termination of the Prior Credit Agreement was recognized as an extinguishment. As of November 19, 2021, the carrying amount of the debt related to the Prior Credit Agreement consisted of principal of $
The Credit Facility is secured by a lien on substantially all of ANI Pharmaceuticals, Inc.’s and its principal domestic subsidiary’s assets and any future domestic subsidiary guarantors’ assets. The Credit Facility is subject to customary financial and nonfinancial covenants.
The carrying value of the current and non-current components of the Term Facility as of December 31, 2021 and Term Loan and Delayed Draw Term Loan under the Prior Credit Agreement as of December 31, 2020 are:
Current | ||||||
December 31, | December 31, | |||||
(in thousands) |
| 2021 |
| 2020 | ||
Current borrowing on debt | $ | |
| $ | | |
Deferred financing costs |
| ( |
| ( | ||
Current debt, net of deferred financing costs | $ | | $ | | ||
Non-Current | ||||||
December 31, | December 31, | |||||
(in thousands) |
| 2021 |
| 2020 | ||
Non-current borrowing on debt | $ | | $ | | ||
Deferred financing costs |
| ( |
| ( | ||
Non-current debt, net of deferred financing costs and current component | $ | | $ | | ||
As of December 31, 2021, we had a $
91
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
in the consolidated balance sheets and $
The contractual maturity of our Term Facility is as follows for the years ending December 31:
(in thousands) |
| Term Facility | ||
2022 | $ | | ||
2023 |
| | ||
2024 |
| | ||
2025 |
| | ||
2026 | | |||
2027 and thereafter | | |||
Total | $ | | ||
The following table sets forth the components of total interest expense related to the Term Facility and the Term Loan, DDTL, and Revolver under our Prior Credit Agreement recognized in our consolidated statements of operations for the year ended December 31:
Years Ended December 31, | |||||||||
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Contractual coupon | $ | | $ | | $ | | |||
Amortization of debt discount |
| — |
| — |
| | |||
Amortization of finance fees |
| |
| |
| | |||
Capitalized interest |
| ( |
| ( |
| ( | |||
$ | | $ | | $ | | ||||
4. DERIVATIVE FINANCIAL INSTRUMENT AND HEDGING ACTIVITY
In April 2020, we entered into an interest rate swap with Citizens Bank, N.A. to manage our exposure to changes in LIBOR-based interest rates underlying total borrowings under term facilities related to our Prior Credit Agreement. The interest rate swap matures in December 2026. Concurrent with the termination of the Prior Credit Agreement and entry into the Credit Agreement with Truist Bank, the interest rate swap with a notional value of $
During the year ended December 31, 2021, the change in fair value of the interest rate swaps was a gain of $
92
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
5. INVENTORIES
Inventories consist of the following as of December 31:
December 31, | December 31, | ||||||
(in thousands) |
| 2021(1) |
| 2020 |
| ||
Raw materials | $ | | $ | | |||
Packaging materials |
| |
| | |||
Work-in-progress |
| |
| | |||
Finished goods |
| |
| | |||
| |
| | ||||
Reserve for excess/obsolete inventories |
| ( |
| ( | |||
Inventories, net | $ | | $ | | |||
| (1) | Includes inventory acquired in the acquisition of Novitium (Note 2). |
6. PROPERTY, PLANT, AND EQUIPMENT
Property, plant, and equipment consist of the following as of December 31:
December 31, | December 31, | |||||
(in thousands) |
| 2021(1) |
| 2020 | ||
Land | $ | | $ | | ||
Buildings |
| |
| | ||
Machinery, furniture, and equipment |
| |
| | ||
Construction in progress |
| |
| | ||
| |
| | |||
Less: accumulated depreciation |
| ( |
| ( | ||
Property and equipment, net | $ | | $ | | ||
| (1) | Includes property and equipment acquired in the acquisition of Novitium (Note 2). |
Depreciation expense for the years ended December 31, 2021, 2020, and 2019 totaled $
7. INTANGIBLE ASSETS
Goodwill
As a result of our 2013 merger with BioSante Pharmaceuticals, Inc., we recorded goodwill of $
For the goodwill impairment analyses performed at October 31, 2021 and 2020, we performed qualitative assessments to determine whether it was more likely than not that our goodwill asset was impaired in order to determine the necessity of performing a quantitative impairment test, under which management would calculate the asset’s fair value. When performing the qualitative assessments, we evaluated events and circumstances that would affect the significant inputs used to determine the fair value of the goodwill. Based on our assessments of the aforementioned factors, it was determined that it was more likely than not that the fair value of our one reporting unit is greater than its carrying amount as of October 31, 2021 and 2020, and therefore no quantitative testing for impairment was required.
In addition to the qualitative impairment analysis performed at October 31, 2021, there were no events or changes in circumstances that would have reduced the fair value of our reporting unit below its carrying value from October 31, 2021 to December 31, 2021.
93
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
2021, 2020, and 2019, and the balance of goodwill was $
Intangible Assets
The components of net definite-lived intangible assets and net indefinite-lived intangible assets other than goodwill are as follows:
December 31, 2021 | December 31, 2020 | Weighted Average | |||||||||||||
Gross Carrying | Accumulated | Gross Carrying | Accumulated | Amortization | |||||||||||
(in thousands) |
| Amount |
| Amortization |
| Amount |
| Amortization |
| Period | |||||
Definite-Lived Intangible Assets: | |||||||||||||||
Acquired ANDA intangible assets | $ | | $ | ( | $ | | $ | ( |
| years | |||||
NDAs and product rights |
| |
| ( |
| |
| ( |
| years | |||||
Marketing and distribution rights |
| |
| ( |
| |
| ( |
| years | |||||
Non-compete agreement |
| |
| ( |
| |
| ( |
| years | |||||
Customer relationships | | ( | — | — | years | ||||||||||
Indefinite-Lived Intangible Assets: | |||||||||||||||
In process research and development | | — | — | — | Indefinite | ||||||||||
Total Intangible Assets, net | $ | | $ | ( | $ | | $ | ( | years | ||||||
Amortization expense was $
Expected future amortization expense is as follows for the years ending December 31:
(in thousands) |
| ||
2022 | $ | | |
2023 |
| | |
2024 |
| | |
2025 |
| | |
2026 |
| | |
2027 and thereafter |
| | |
Total | $ | |
Expected amortization expense is an estimate. Actual amounts of amortization expense may differ due to timing of regulatory approvals related to IPR&D assets, additional intangible assets acquired, impairment of intangible assets, and other events.
8. FAIR VALUE DISCLOSURES
Fair value is the price that would be received from the sale of an asset or paid to transfer a liability assuming an orderly transaction in the most advantageous market at the measurement date. U.S. GAAP establishes a hierarchical disclosure framework which prioritizes and ranks the level of observability of inputs used in measuring fair value.
The inputs used in measuring the fair value of cash and cash equivalents are considered to be Level 1 in accordance with the three-tier fair value hierarchy. The fair market values are based on period-end statements supplied by the various banks and brokers that held the majority of our funds. The fair value of short-term financial instruments (primarily accounts receivable, prepaid expenses, accounts payable, accrued expenses, and other current liabilities) approximate their carrying values because of their short-term nature. The Term Facility bears an interest rate that fluctuates with the changes in LIBOR and, because the variable interest rates approximate market borrowing rates available to us, we believe the carrying values of these borrowings approximated their fair values at December 31, 2021 and 2020.
94
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
Contingent Value Rights
Our contingent value rights (“CVRs”), which were granted coincident with our merger with BioSante Pharmaceuticals, Inc. and expire in June 2023, are considered to be contingent consideration and are classified as liabilities. As such, the CVRs were recorded as purchase consideration at their estimated fair value, using Level 3 inputs, and are marked to market each reporting period until settlement. The fair value of CVRs is estimated using the present value of management’s projection of the expected payments pursuant to the terms of the CVR agreement, which is the primary unobservable input. If our projection or expected payments were to increase substantially, the value of the CVRs could increase as a result. The present value of the liability was calculated using a discount rate of
Interest Rate Swap
The fair value of our interest rate swap is estimated based on the present value of projected future cash flows using the LIBOR forward rate curve. The model used to value the interest rate swap includes inputs of readily observable market data, a Level 2 input. As described in detail in Note 4, the fair value of the interest rate swap was a $
Contingent Consideration
In connection with the acquisition of Novitium, we may pay up to $
The discounted cash flow method used to value this contingent consideration includes inputs of not readily observable market data, which are Level 3 inputs. As of the November 19, 2021 acquisition date, the contingent consideration had a fair value of $
The recurring Level 3 fair value measurements of contingent consideration for which a liability is recorded include the following significant unobservable inputs:
Payment Type | Valuation Technique | Unobservable Input | Range | |||||
Profit-based milestone payments | Probability-weighted discounted cash flow | Discount rate | ||||||
Projected fiscal year of payment | 2023-2029 | |||||||
Product development-based milestone payments | Probability-weighted discounted cash flow | Discount rate | ||||||
Probability of payment | ||||||||
Projected fiscal year of payment | 2023-2024 |
95
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
The following table presents our financial assets and liabilities accounted for at fair value on a recurring basis as of December 31, 2021 and December 31, 2020, by level within the fair value hierarchy:
(in thousands) | Fair Value at | |||||||||||
Description | December 31, 2021 | Level 1 | Level 2 | Level 3 | ||||||||
Liabilities |
|
|
|
|
|
|
|
| ||||
Contingent consideration | $ | | $ | — | $ | — | $ | | ||||
Interest rate swaps | $ | | $ | — | $ | | $ | — | ||||
CVRs | $ | — | $ | — | $ | — | $ | — | ||||
| Fair Value at |
|
|
| ||||||||
Description | December 31, 2020 | Level 1 | Level 2 | Level 3 | ||||||||
Liabilities |
|
|
|
|
|
|
|
| ||||
Contingent consideration | $ | — | $ | — | $ | — | $ | — | ||||
Interest rate swaps | $ | | $ | — | $ | | $ | — | ||||
CVRs | $ | — | $ | — | $ | — | $ | — | ||||
Financial Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
We have no financial assets and liabilities that are measured at fair value on a non-recurring basis.
Non-Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
We have no non-financial assets and liabilities that are measured at fair value on a recurring basis.
Non-Financial Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
We measure our long-lived assets, including property, plant, and equipment, ROU assets, intangible assets, and goodwill, at fair value on a non-recurring basis. These assets are recognized at fair value when they are deemed to be other-than-temporarily impaired. During the year ended December 31, 2021, we recognized an impairment charge of $
Acquired Non-Financial Assets Measured at Fair Value
In April 2021, we acquired three NDAs and an ANDA and certain related inventories from Sandoz, Inc. for total consideration of $
In July 2020, we acquired an ANDA and certain related inventories from a private company for total consideration of $
96
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
inputs. The ANDA was being amortized in full over its useful life of
In January 2020, we completed the acquisition of the U.S. portfolio of
9. MEZZANINE AND STOCKHOLDERS’ EQUITY
Stockholders’ Equity
Authorized shares
We are authorized to issue up to
There were
There were
97
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Mezzanine Equity
PIPE Shares
Concurrently with the execution of the Agreement and Plan of Merger, and as financing for a portion of the acquisition, on March 8, 2021, we entered into an Equity Commitment and Investment Agreement with Ampersand (the “PIPE Investor”), pursuant to which we agreed to issue and sell to the PIPE Investor, and the PIPE Investor agreed to purchase,
The PIPE Shares accrue dividends at
In case of a liquidation event, the holder of the PIPE Shares will be entitled to receive, in preference to holders of our common stock, the greater of (i) the PIPE Shares’ purchase price plus any accrued and unpaid dividends thereon and (ii) the amount the holder of the PIPE Shares would have received in the liquidation event if it had converted its PIPE Shares into our common stock. The PIPE Shares will have voting rights, voting as one series with our common stock, on as-converted basis, and will have separate voting rights on any (i) amendment to the Certificate of Designation of Preferences, Rights and Limitations of Series A Convertible Preferred Stock (the “Certificate”) that adversely amends and relates solely to the terms of the PIPE Shares and (ii) issuance of additional series A convertible preferred stock. In case of a change of control of ANI, the PIPE Shares will be redeemed at the greater of (i) the PIPE Shares’ purchase price plus any accrued and unpaid dividends thereon and (ii) the change of control transaction consideration that the holder of the PIPE Shares would have received if it had converted into our common stock.
There were
10. STOCK-BASED COMPENSATION
Employee Stock Purchase Plan
In July 2016, we commenced administration of the ANI Pharmaceuticals, Inc. 2016 Employee Stock Purchase Plan. The Board of Directors and shareholders approved a maximum of
98
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
The following table summarizes ESPP expense incurred under the 2016 Employee Stock Purchase Plan and included in our consolidated statements of operations:
(in thousands) | Years Ended December 31, | ||||||||
| 2021 |
| 2020 |
| 2019 | ||||
Cost of sales | $ | | $ | | $ | | |||
Research and development |
| |
| |
| | |||
Selling, general, and administrative |
| |
| |
| | |||
$ | | $ | | $ | | ||||
Stock Incentive Plan
Equity-based service awards are granted under the ANI Pharmaceuticals, Inc. Amended and Restated 2008 Stock Incentive Plan (the “2008 Plan”). As of December 31, 2021,
From time to time, we may grant stock options to employees through an inducement grant outside of our 2008 Plan to induce prospective employees to accept employment with us (the “Inducement Grants”). The options are granted at an exercise price equal to the fair market value of a share of our common stock on the respective grant date and are generally exercisable in four equal annual installments beginning on the first anniversary of the respective grant date. The grants are made pursuant to inducement grants outside of our stockholder approved equity plan as permitted under the Nasdaq Stock Market listing rules.
We measure the cost of equity-based service awards based on the grant-date fair value of the award. The cost is recognized ratably over the period during which an employee is required to provide service in exchange for the award or the requisite service period. We recognize stock-based compensation expense ratably over the vesting periods of the awards.
The following table summarizes stock-based compensation expense incurred under the 2008 Plan and Inducement Grant and included in our consolidated statements of operations:
Years Ended December 31, | |||||||||
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Cost of sales | $ | | $ | | $ | | |||
Research and development |
| |
| |
| | |||
Selling, general, and administrative |
| |
| |
| | |||
$ | | $ | | $ | | ||||
We recognized income tax benefits of $
Stock Options
Outstanding stock options granted to employees and consultants generally vest over a period of
99
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
For 2021, 2020, and 2019, the fair value of each option grant was estimated using the Black-Scholes option-pricing model, using the following assumptions:
Years Ended December 31, | ||||||
| 2021 |
| 2020 |
| 2019 | |
Expected option life (years) | ||||||
Risk-free interest rate |
|
|
| |||
Expected stock price volatility |
|
|
| |||
Dividend yield |
|
| ||||
We use the simplified method to estimate the expected option life of options. The risk-free interest rate used is the yield on a U.S. Treasury note as of the grant date with a maturity equal to the estimated life of the option. We calculated an estimated volatility rate based on our historical stock price. We have not issued a cash dividend on our common shares in the past nor do we have any current plans to do so in the future; therefore, an expected dividend yield of
A summary of stock option activity under the 2008 Plan and Inducement Grants during the years ended December 31, 2021, 2020, and 2019 is presented below:
|
|
| Weighted |
| Weighted |
| ||||||||
Average | Average | |||||||||||||
Weighted | Grant- | Remaining | ||||||||||||
(in thousands, except per share and | Option | Average | date | Term | Aggregate | |||||||||
remaining term data) | Shares | Exercise Price | Fair Value | (years) | Intrinsic Value | |||||||||
Outstanding December 31, 2018 |
| | $ | |
| $ | | |||||||
Granted |
| |
| | $ | | ||||||||
Exercised |
| ( |
| |
| | ||||||||
Forfeited |
| ( |
| | ||||||||||
Expired |
| ( |
| | ||||||||||
Outstanding December 31, 2019 |
| | $ | |
| $ | | |||||||
Granted |
| |
| | $ | | ||||||||
Exercised |
| ( |
| |
| | ||||||||
Forfeited |
| ( |
| | ||||||||||
Expired |
| — |
| — | ||||||||||
Outstanding at December 31, 2020 |
| | $ | |
| $ | | |||||||
Granted |
| |
| | $ | | ||||||||
Exercised |
| ( |
| |
| | ||||||||
Forfeited |
| ( |
| | ||||||||||
Expired |
| ( |
| | ||||||||||
Outstanding at December 31, 2021 |
| | $ | |
| $ | | |||||||
Exercisable at December 31, 2021 |
| | $ | |
| $ | | |||||||
As of December 31, 2021, there was $
100
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Restricted Stock Awards
Restricted stock awards (“RSAs”) granted to employees generally vest over a period of
Shares of our common stock delivered to employees and directors will be unrestricted upon vesting. During the vesting period, the recipient of the restricted stock has full voting rights as a stockholder and would receive dividends, if declared, even though the restricted stock remains subject to transfer restrictions and will generally be forfeited upon termination of the officer prior to vesting. The fair value of each RSA is based on the market value of our stock on the date of grant.
A summary of RSA activity under the Plan during the years ended December 31, 2021, 2020, and 2019 is presented below:
|
| Weighted |
| ||||
Average Grant | Weighted Average | ||||||
(in thousands, except per share and | Date Fair | Remaining Term | |||||
remaining term data) | Shares | Value | (years) | ||||
Unvested at December 31, 2018 |
| | $ | |
| ||
Granted |
| |
| |
|
| |
Vested |
| ( |
| |
|
| |
Forfeited |
| ( |
| |
|
| |
Unvested at December 31, 2019 |
| | $ | |
| ||
Granted |
| |
| |
|
| |
Vested |
| ( |
| |
|
| |
Forfeited |
| ( |
| |
|
| |
Unvested at December 31, 2020 |
| | $ | |
| ||
Granted |
| |
| |
|
| |
Vested |
| ( |
| |
|
| |
Forfeited |
| ( |
| |
|
| |
Unvested at December 31, 2021 |
| | $ | |
| ||
As of December 31, 2021, there was $
11. INCOME TAXES
On August 6, 2018, ANI Pharmaceuticals Canada Inc. (“ANI Canada”) acquired all the issued and outstanding equity interests of WellSpring in a non-taxable transaction. Following the consummation of the transaction, WellSpring was merged into ANI Canada. For U.S. Federal and state income tax purposes, ANI Canada is not part of ANI’s consolidated group; rather, ANI Canada is subject to income taxes only in Canada and solely based on its stand-alone operations. The foreign current and foreign deferred provisions (benefits) below represent our tax provision (benefit) from the Canadian, Indian, and Israeli taxing jurisdictions.
We are required to establish a valuation allowance for deferred tax assets if, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. We consider the projected future taxable income and tax planning strategies in making this assessment.
As part of purchase accounting in 2018, we established net deferred tax assets relating to differences in the book bases (determined based on fair value purchase accounting) and tax bases (determined based on the carryover nature of the nontaxable transaction) of ANI Canada’s assets and liabilities of approximately $
101
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
not be realized. During 2019, we adopted an intercompany transfer pricing policy that uses the “comparable profits method” for pricing intercompany services between ANI Pharmaceuticals, Inc. and ANI Canada. For U.S. and Canadian tax purposes, the policy was adopted in conjunction with the acquisition date of August 6, 2018. As a result of the newly adopted transfer pricing policy, our assessment of the amount of ANI Canada’s deferred tax assets that are more likely than not to be realized changed and, as a result, during 2019, we released the remaining net valuation allowance related to ANI Canada’s deferred tax assets.
As of December 31, 2021 and 2020, our consolidated valuation allowance was $
Our total provision for income taxes consists of the following for the years ended December 31, 2021, 2020, and 2019:
(in thousands) |
| 2021 |
| 2020 |
| 2019 | |||
Current income tax provision: |
|
|
|
|
|
| |||
Federal | $ | | $ | | $ | | |||
State |
| |
| |
| | |||
Foreign |
| |
| — |
| — | |||
Total |
| |
| |
| | |||
Deferred income tax benefit |
|
|
|
|
|
| |||
Federal |
| ( |
| ( |
| ( | |||
State |
| ( |
| |
| ( | |||
Foreign |
| |
| |
| | |||
Total |
| ( |
| ( |
| ( | |||
Change in valuation allowance |
| |
| ( |
| ( | |||
Total benefit for income taxes | $ | ( | $ | ( | $ | ( | |||
The difference between our expected income tax provision from applying U.S. Federal statutory tax rates to the pre-tax income and actual income tax provision relates primarily to the effect of the following:
As of December 31, |
| ||||||
| 2021 |
| 2020 |
| 2019 |
| |
US Federal statutory rate |
| | % | | % | | % |
State taxes, net of Federal benefit |
| | % | | % | | % |
Foreign taxes |
| ( | % | ( | % | | % |
Change in valuation allowance |
| ( | % | | % | ( | % |
Stock-based compensation |
| ( | % | ( | % | ( | % |
Non-deductible costs | ( | % | ( | % | | % | |
Change in state apportionment factors, state and foreign rates | | % | ( | % | ( | % | |
Research and experimentation and charitable credits | | % | | % | ( | % | |
Transfer pricing and other | ( | % | | % | ( | % | |
Effective income tax rate |
| | % | | % | ( | % |
102
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Deferred income taxes reflect the net tax effects of differences between the bases of assets and liabilities for financial reporting and income tax purposes. Our deferred income tax assets and liabilities consisted of the following:
As of December 31, | ||||||
(in thousands) |
| 2021 |
| 2020 | ||
Deferred tax assets: |
|
|
|
| ||
Accruals and advances | $ | | $ | | ||
Stock-based compensation | | | ||||
Accruals for chargebacks and returns |
| |
| | ||
Inventory |
| |
| | ||
Intangible asset |
| |
| | ||
Net operating loss carryforwards |
| |
| | ||
Other |
| |
| | ||
Total deferred tax assets | $ | | $ | | ||
Deferred tax liabilities: |
|
|
|
| ||
Depreciation | $ | ( | $ | ( | ||
Intangible assets |
| ( |
| ( | ||
Other |
| ( |
| ( | ||
Total deferred tax liabilities | $ | ( | $ | ( | ||
Valuation allowance |
| ( |
| ( | ||
Deferred tax assets, net of deferred tax liabilities and valuation allowance | $ | | $ | | ||
As of December 31, 2021, we had U.S. federal net operating loss carryforwards of approximately $
We are subject to income taxes in numerous jurisdictions in the U.S., Canada, and India. Significant judgment is required in evaluating our tax positions and determining our provision for income taxes. We establish liabilities for tax-related uncertainties based on estimates of whether, and the extent to which, additional taxes will be due. These liabilities are established when we believe that certain positions might be challenged despite our belief that our tax return positions are fully supportable. We adjust these liabilities in light of changing facts and circumstances, such as the outcome of a tax audit. The provision for income taxes includes the impact of changes to the liability that is considered appropriate. We identified no material uncertain income tax positions as of December 31, 2021 and 2020.
We are subject to income tax audits in all jurisdictions for which we file tax returns. Tax audits by their nature are often complex and can require several years to complete. All of our income tax returns remain subject to examination by tax authorities due to the availability of net operating loss carryforwards.
12. COMMITMENTS AND CONTINGENCIES
Operating Leases
All our existing leases as of December 31, 2021 are classified as operating leases. As of December 31, 2021, we have twelve material operating leases for facilities and office equipment with remaining terms expiring from 2022 through 2026 and a weighted average remaining lease term of
103
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Rent expense for the years ended December 31, 2021 and 2020 consisted of the following:
| Year Ended December 31, | |||||
(in thousands) |
| 2021 | 2020 | |||
Operating lease costs | $ | | $ | | ||
Variable lease costs |
| |
| | ||
Total lease costs | $ | | $ | | ||
A maturity analysis of our operating leases follows:
(in thousands) |
| ||
Future payments: | |||
2022 | $ | | |
2023 |
| | |
2024 |
| | |
2025 |
| | |
2026 and thereafter |
| | |
Total | $ | | |
Discount | ( | ||
| |||
Current lease liability | ( | ||
$ | |
Vendor Purchase Minimums
We have supply agreements with three vendors that include purchase minimums. Pursuant to these agreements, we will be required to purchase a total of $
Government Regulation
Our products and facilities are subject to regulation by a number of federal and state governmental agencies, such as the Drug Enforcement Administration (“DEA”), the Food and Drug Administration (“FDA”), the Centers for Medicare and Medicaid Services (“CMS”), Health Canada, the Central Drugs Standard Control Organization (“CDSCO”), The Narcotics Control Bureau (“NCB”), and India’s Ministry of Health and Family Welfare (“MoHFW”). The FDA, in particular, maintains oversight of the formulation, manufacture, distribution, packaging, and labeling of all of our products. The DEA, Health Canada, and NCB maintain oversight over our products that are considered controlled substances.
Unapproved Products
Two of our products, Esterified Estrogen with Methyltestosterone (“EEMT”) and Opium Tincture, are marketed without approved NDAs or ANDAs. During the years ended December 31, 2021, 2020, and 2019, net revenues for these products totaled $
The FDA's policy with respect to the continued marketing of unapproved products appears in the FDA's September 2011 Compliance Policy Guide Sec. 440.100 titled “Marketed New Drugs without Approved NDAs or ANDAs.” Under this policy, the FDA has stated that it will follow a risk-based approach with regard to enforcement against marketing of unapproved products. The FDA evaluates whether to initiate enforcement action on a case-by-case basis, but gives higher priority to enforcement action against products in certain categories, such as those with potential safety risks or that lack evidence of effectiveness.
We continue to believe that, so long as we comply with applicable manufacturing standards, the FDA will continue to operate on a risk-based approach and will not take action against us. However, we can offer no assurance
104
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
that the FDA will continue to follow this approach or that it will not take a contrary position with any individual product or group of products. If the FDA were to move away from the risk-based approach to enforcement against marketing of unapproved products, we may be required to seek FDA approval for these products or withdraw such products from the market. If we decide to withdraw the products from the market, our net revenues for generic pharmaceutical products would decline materially, and if we decide to seek FDA approval, we would face increased expenses and might need to suspend sales of the products until such approval was obtained, and there are no assurances that we would receive such approval.
In addition, one group of products that we manufacture on behalf of a contract customer is marketed by that customer without an approved NDA. If the FDA took enforcement action against such customer, the customer may be required to seek FDA approval for the group of products or withdraw them from the market. Our contract manufacturing revenues for the group of unapproved products for the years ended December 31, 2021, 2020, and 2019 were $
Legal proceedings
We are involved, and from time to time may become involved, in various disputes, governmental and/or regulatory inquiries, investigations, government reimbursement related actions and litigation. These matters are complex and subject to significant uncertainties. As such, we cannot accurately predict the outcome, or the effects of the legal proceedings described below. While we believe that we have valid claims and/or defenses in the litigation and other matters described below, litigation is inherently unpredictable, and the outcome of the proceedings could result in losses, including substantial damages, fines, civil or criminal penalties and injunctive or administrative remedies. We intend to vigorously prosecute and/or defend these matters, as appropriate, however, from time to time, we may settle or otherwise resolve these matters on terms and conditions that we believe are in our best interests. Resolution of any or all claims, investigations, and legal proceedings, individually or in the aggregate, could have a material adverse effect on our results of operations and/or cash flows in any given accounting period or on our overall financial condition.
Some of these matters with which we are involved are described below, and unless otherwise disclosed, we are unable to predict the outcome of the matter or to provide an estimate of the range of reasonably possible material losses. We record accruals for loss contingencies to the extent we conclude it is probable that a liability has been incurred and the amount of the loss can be reasonably estimated.
From time to time, we are also involved in other pending proceedings for which, in our opinion based upon facts and circumstances known at the time, either the likelihood of loss is remote or any reasonably possible loss associated with the resolution of such proceedings is not expected to be material to our results, and therefore remain undisclosed. If and when any reasonably possible losses associated with the resolution of such other pending proceedings, in our opinion, become material, we will disclose such matters.
Furthermore, like all pharmaceutical manufacturers, we are periodically exposed to product liability claims. The prevalence of these claims could limit our coverage under future insurance policies or cause those policies to become more expensive, which could harm our business, financial condition, and operating results. Recent trends in the product liability and director and officer insurance markets is to exclude matters related to certain classes of drugs. Our policies have been subject to such exclusions which place further potential risk of financial loss on us.
Legal fees for litigation-related matters are expensed as incurred and included in the consolidated statements of operations under the selling, general, and administrative expense line item.
Commercial Litigation
In November of 2017, we were served with a complaint filed by Arbor Pharmaceuticals, LLC, in the United States District Court for the District of Minnesota. The complaint alleged false advertising and unfair competition in violation of Section 43(a) of the Lanham Act, Section 1125(a) of Title 15 of the United States Code, and Minnesota State law, under the premise that we sold an unapproved Erythromycin Ethylsuccinate (“EES”) product during the period between September 27, 2016 and November 2, 2018. The complaint sought a trial by jury and monetary
105
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
damages (inclusive of actual and consequential damages, treble damages, disgorgement of ANI profits, and legal fees) of an unspecified amount. Discovery in this action closed on March 31, 2019 and trial was scheduled to commence on August 25, 2021. On August 3, 2021, the Company entered into a Settlement Agreement with Arbor Pharmaceuticals, LLC to resolve all claims related to Civil Action 17-4910, Arbor Pharmaceuticals, LLC (“Arbor”) v. ANI Pharmaceuticals, Inc., which was pending trial in the United States District Court for the District of Minnesota. Under the terms of the agreement, ANI paid Arbor $
On December 3, 2020, class action complaints were filed against the Company on behalf of putative classes of direct and indirect purchasers of the drug Bystolic. On December 23, 2020, six individual purchasers of Bystolic, CVS, Rite Aid, Walgreen, Kroger, Albertsons, and H-E-B, filed complaints against the Company. On March 15, 2021, the plaintiffs in these actions filed amended complaints. All amended complaints are substantively identical. The plaintiffs in these actions allege that, beginning in 2012, Forest Laboratories, the manufacturer of Bystolic, entered into anticompetitive agreements when settling patent litigation related to Bystolic with seven potential manufacturers of a generic version of Bystolic: Hetero, Torrent, Alkem/Indchemie, Glenmark, Amerigen, Watson, and various of their corporate parents, successors, subsidiaries, and affiliates. ANI itself was not a party to patent litigation with Forest concerning Bystolic and did not settle patent litigation with Forest. The plaintiffs named the Company as a defendant based on the Company’s January 8, 2020 Asset Purchase Agreement with Amerigen. The complaints alleged that the 2013 patent litigation settlement agreement between Forest and Amerigen violated federal and state antitrust laws and state consumer protection laws by delaying the market entry of generic versions of Bystolic. Plaintiffs alleged they paid higher prices as a result of delayed generic competition. Plaintiffs sought damages, trebled or otherwise multiplied under applicable law, injunctive relief, litigation costs and attorneys’ fees. The complaints did not specify the amount of damages sought from the Company or other defendants and the Company at this early stage of the litigation cannot reasonably estimate the potential damages that the plaintiffs will seek. The cases have been consolidated in the United States District Court for the Southern District of New York as In re Bystolic Antitrust Litigation, Case No. 20-cv-005735 (LJL). On April 23, 2021, the Company and other defendants filed motions to dismiss the amended complaints. On January 24, 2022, the court dismissed all claims brought by the plaintiffs without prejudice. The court granted the plaintiffs until February 22, 2022 to file amended complaints, which were filed on that date. The newly amended complaints contain substantially similar claims. The Company disputes any liability in these matters.
On March 24, 2021, Azurity Pharmaceuticals, Inc. (“Azurity”) filed a complaint in the United States District Court for the District of Minnesota against ANI Pharmaceuticals, Inc., asserting that ANI’s vancomycin hydrochloride oral solution drug product infringes U.S. Patent No. 10,688,046. The complaint sought injunctive relief, damages, including lost profits and/or royalty, treble damages, and attorneys’ fee and costs. On February 15, 2022, the Company entered into a settlement agreement with Azurity to resolve all claims related to this action. Under the terms of the agreement, Azurity granted ANI a non-exclusive, non-transferable, non-sublicensable, royalty-bearing license under its Patents to sell ANI product in the United States and dismissed the action with prejudice. In exchange, we paid Azurity $
On April 1, 2021, United Therapeutics Corp. and Supernus Pharmaceuticals, Inc. (“UTC/Supernus”) filed a complaint in the United States District Court for the District of Delaware against ANI Pharmaceuticals, Inc., asserting that ANI's proposed Treprostinil extended release drug product, which is subject to ANI’s Abbreviated New Drug Application No. 215667, infringes U.S. Patent Nos. 7,417,070, 7,544,713, 8,252,839, 8,349,892, 8,410,169, 8,747,897, 9,050,311, 9,278,901, 9,393,203, 9,422,223, 9,593,066 and 9,604,901 (“the Asserted Patents”). The complaint seeks injunctive relief, attorneys' fee and costs. ANI filed its answer and counterclaims on May 28, 2021, denying UTC/Supernus’ allegations and seeking declaratory judgment that ANI has not infringed any valid and enforceable claim of the Asserted Patents, that the Asserted Patents are invalid, and an award of attorneys’ fees and costs. Trial is set for May 8, 2023.
106
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
Industry Related Litigation
In July 2020, ANI and Novitium were served with a complaint brought by the Office of the Attorney General of the State of New Mexico against manufacturers and sellers of ranitidine products. The complaint asserts a public nuisance claim and a negligence claim against the generic ranitidine manufacturer defendants, including ANI and Novitium. The public nuisance claim asserts that the widespread sale of ranitidine products in the state created a public nuisance that requires a state-wide medical monitoring program of New Mexico residents for the development of colorectal cancer, stomach cancer, gastrointestinal disorders and liver disease. As damages, New Mexico asks that the defendants fund this medical monitoring program. The negligence claims assert that the defendants were negligent in selling the product, essentially alleging that it was unreasonable to have the product on the market. With respect to that claim, New Mexico asserts that it paid for ranitidine products through state-funded insurance and health-care programs. On December 15, 2020, the case was removed to federal court and transferred to the In re Zantac multidistrict litigation (“MDL”) pending in the United States District Court for the Southern District of Florida. New Mexico moved for remand to state court. The MDL court granted the remand motion on February 25, 2021. On April 16, 2021, New Mexico filed an amended complaint in the New Mexico First Judicial District Court in Santa Fe County. It did not name ANI in the amended complaint, effectively voluntarily dismissing ANI from the action. Novitium is named as a Defendant in the amended complaint. According to Novitium’s records, Novitium sold approximately 42 bottles of ranitidine indirectly into New Mexico, and received no funds from any state funded health care plan or Medicaid. The Defendants filed a motion to dismiss the claims asserted in the New Mexico litigation based primarily on preemption. The motion was denied in August 2021.
In December 2020, the City of Baltimore served ANI and Novitium with a complaint against manufacturers and sellers of ranitidine products. The City of Baltimore complaint tracks the allegations of the New Mexico complaint. The Baltimore action was removed to federal court and transferred to the In re Zantac MDL on February 1, 2021. The City of Baltimore moved for remand, which was granted on April 1, 2021. The parties stipulated to allow the City of Baltimore to file an amended complaint in the Circuit Court of Maryland for Baltimore City in “due course,” without a specific filing deadline. On June 23, 2021, the City of Baltimore filed an amended complaint. The City of Baltimore did not name ANI in its amended complaint, effectively voluntarily dismissing ANI from the action. Novitium was named as a defendant in the amended complaint. Defendants in the Baltimore action filed a motion to dismiss on based primarily on preemption to which Novitium joined. The motion was granted as to all generic manufacturer defendants on January 28, 2022, and all claims against Novitium were dismissed with prejudice. The deadline for the City to file an appeal was February 28, 2022.
ANI and Novitium dispute any liability in these matters.
Product Liability Related Litigation
All manufacturers of the drug Reglan and its generic equivalent metoclopramide, including ANI, have faced allegations from plaintiffs in various states claiming bodily injuries as a result of ingestion of metoclopramide or its brand name, Reglan, prior to the FDA’s February 2009 Black Box warning requirement (“legacy claims”). All these original legacy claims were settled or closed out, including a series of claims in California that were resolved by coordinated proceeding and settlement. Our insurance company assumed the defense of the legacy claims and paid all losses in settlement of the California legacy claims. In March 2019, we were served with a lawsuit in the Superior Court of California, County of Riverside, adding us as a defendant in a complaint filed in July 2017 that is alleged not to have been part of the original settled legacy claims. This new claim was dismissed with prejudice in July 2021 and the matter is now closed.
In June 2020, ANI was served with a personal injury complaint in the case of Koepsel v. Boehringer Ingelheim Pharmaceuticals, et al., MDL No. 20-MD-2924, Case No. 9:20-cv-80882-RLR, filed in the United States District Court for Southern District of Florida, in which the plaintiff alleges that he developed kidney cancer in 2018 as a result of taking over the counter medication containing ranitidine. The Koepsel action was filed within an existing multidistrict litigation concerning ranitidine-containing drugs pending in the Southern District of Florida before Judge Robin L. Rosenberg, In re Zantac MDL, 20 MDL 2924. A Master Personal Injury Complaint (“MPIC”) in that MDL that was filed on June 22, 2020 also named ANI and Novitium as defendants. ANI was dismissed from the Koepsel case on August 21, 2020 and was dismissed from the MPIC on September 8, 2020. On December 31, 2020,
107
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
after ANI was dismissed, the district court dismissed the MPIC claims against generic manufacturer defendants partially with prejudice and partially with leave to replead. The failure to warn and design defect claims were dismissed with prejudice on preemption grounds. An Amended Master Personal Injury Complaint was filed on February 8, 2021, which did not name ANI but did name Novitium. By opinion dated July 8, 2021, the district court dismissed all claims against the generic manufacturer defendants with prejudice on preemption grounds. That decision is on appeal to the Eleventh Circuit Court of Appeals.
ANI and Novitium were named in other individual personal injury complaints filed in MDL 20 MD 2924 in which plaintiffs allege that they developed cancer after taking prescription and over the counter medication containing ranitidine. ANI was served with complaints in five of those additional cases: Cooper v. Boehringer Ingelheim Pharmaceuticals, et al., MDL No. 20-MD-2924, Case No. 9:20-cv-81130-RLR (served September 30, 2020), Lineberry v. Amneal Pharmaceuticals, LLC, et al., MDL No. 20-MD-2924, Case No. 9:20-cv-81079-RLR (served August 20, 2020), Lovette v. Amneal Pharmaceuticals, LLC, et al., MDL No. 20-MD-2924, Case No. 9:20-cv-81040-RLR (served August 26, 2020), Hightower v. Pfizer, et al, MDL No. 20-MD-2924, Case No. 9-20-cv-82214-RLR (served December 16, 2020) and Bird v. Boehringer Ingelheim Pharmaceuticals, et al., MDL No. 20-MD-2924, Case No. 9-20-cv-80837-RLR (served December 30, 2020). We have informed counsel for the plaintiffs that ANI did not sell an over the counter ranitidine product and sold a generic prescription ranitidine product for a limited two-month period of time, from July 2019 to September 2019. ANI’s product was voluntarily recalled in January 2020. Each of the plaintiffs in the five pending cases alleges a cancer diagnosis prior to the time that ANI sold ranitidine, and we have informally sought dismissal from these cases on that basis. ANI was voluntarily dismissed from the Cooper, Lineberry and Lovette actions on November 20, 2020. ANI was voluntarily dismissed from the Bird action on March 15, 2021 and from the Hightower action on March 29, 2021.
Novitium has been named in 150 short form complaints filed by claimants in the MDL. Those complaints were effectively dismissed with prejudice with the MPIC on July 8, 2021. Counsel for the plaintiffs have been notified that Novitium did not sell an over the counter ranitidine product and sold a generic prescription ranitidine product for a limited period of time, from December 2019 until September 2019. Novitium’s product was voluntarily recalled in October 2019. Out of the 150 claimants, approximately 109 claimants either were diagnosed with cancer before Novitium began manufacturing the product, only took over the counter ranitidine, or took ranitidine before Novitium began manufacturing it. In light of the Court’s dismissal of all claims with prejudice, Novitium has not pursued dismissal of the short form complaints against it at this time.
On February 3, 2022, a complaint was filed in Cook County, Illinois, naming Novitium as a Defendant. The complaint incorrectly identifies Novitium as a “repackager.” The case is styled Ross v. Boehringer Ingelheim Pharmaceuticals, Inc., et. al. The complaint has not yet been served on Novitium. The complaint asserts claims of strict liability/failure to warn, strict liability/design defect, negligent failure to warn, negligent product design, general negligence, negligent misrepresentation, breach of express and implied warranties, and unjust enrichment. At this point, there is no indication that the Plaintiff used a Novitium product.
ANI and Novitium dispute any liability in these MDL matters.
Other Industry Related Matters
On or about September 20, 2017, the Company and certain of its employees were served with search warrants and/or grand jury subpoenas to produce documents and possibly testify relating to a federal investigation of the
generic pharmaceutical industry. We have been cooperating and intend to continue cooperating with the investigation. However, no assurance can be given as to the timing or outcome of the investigation.
13. PURIFIED CORTROPHIN PRE-LAUNCH CHARGES
In January 2016, we acquired the right, title and interest in the NDAs for Cortrophin Gel and Cortrophin-Zinc. Subsequently, we assembled a Cortrophin Gel re-commercialization team of scientists, executed a long-term supply agreement with a supplier of pig pituitary glands, our primary raw material for corticotrophin API, executed a long-term supply agreement with an API manufacturer, with whom we have advanced the manufacture of corticotropin
108
ANI Pharmaceuticals, Inc. and Subsidiaries
Notes to the Consolidated Financial Statements
For the years ended December 31, 2021, 2020, and 2019
API via manufacture of commercial-scale batches, and executed a long-term commercial supply agreement with a current good manufacturing practice (“cGMP”) aseptic fill contract manufacturer.
Prior to the third quarter 2019, all purchases of material, including pig pituitary glands and API, related to the re-commercialization efforts were consumed in research and development activities and recognized as research and development expense in the period in which they were incurred. In the third quarter of 2019, we began purchasing materials that are intended to be used commercially in anticipation of FDA approval of Cortrophin Gel and the resultant product launch. The FDA granted approval of the sNDA of this product on October 29, 2021. Prior to FDA approval, under U.S. GAAP, we were prohibited from capitalizing these pre-launch purchases of materials as inventory, and accordingly, they were charged to expense in the period in which they were incurred. Subsequent to approval, these purchases are recorded as inventory at net realizable value. During the years ended December 31, 2021, 2020, and 2019, we incurred $
14. RELATED PARTY TRANSACTIONS
On March 8, 2021, we entered into an Equity Commitment and Investment Agreement with Ampersand 2020 Limited Partnership, pursuant to which we agreed to issue and sell
In August 2020, we appointed Jeanne Thoma as a director of the Company. Ms. Thoma is the former Chief Executive Officer of SPI Pharmaceuticals, Inc. (“SPI”), who retired in October 2020. SPI supplies ingredients to the Company. We made payments totaling approximately $
In connection with our acquisition of Novitium, we entered into employment agreements with the two executives and founders of Novitium, Muthusamy Shanmugam and Chad Gassert. Both will serve as executive officers of the Company and Mr. Shanmugam was also appointed to the board of directors. Mr. Shanmugam holds a minority interest in Scitus Pharma Services (“Scitus”), which provides clinical research services to Novitium, majority interest in SS Pharma LLC (“SS Pharma”), which acquires and supplies API to Novitium, majority interest in Esjay Pharma LLC (“Esjay”), which provides research and development and facilities consulting services, and a minority interest in Nuray Chemical Private Limited (“Nuray”), which manufactures and supplies API to Novitium. Mr. Gassert holds a minority interest in Scitus. During the period from November 19, 2021 and December 31, 2021, subsequent to our acquisition of Novitium, we paid Esjay an immaterial amount and paid Nuray $
15. SUBSEQUENT EVENT
On February 15, 2022, we settled an outstanding litigation with Azurity Pharmaceuticals, Inc. (“Azurity”). Refer to Note 12 for more information.
109
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized, and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed under the Exchange Act is accumulated and communicated to management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Our management has carried out an evaluation, under the supervision and with the participation of our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act), as of December 31, 2021. Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of the period covered by this report, our disclosure controls and procedures were effective. In designing and evaluating our disclosure controls and procedures, we recognize that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act as a process designed by, or under the supervision of, a company’s principal executive and principal financial officers and effected by a company’s board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP. Our internal control over financial reporting includes those policies and procedures that:
| ● | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect transactions and dispositions of its assets; |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. GAAP, and that receipts and expenditures are being made only in accordance with authorizations of management and directors; and |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of assets that could have a material effect on our consolidated financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of December 31, 2021. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control — Integrated Framework (2013).Based on this assessment, our management has concluded that, as of December 31, 2021, our internal control over financial reporting is effective based on those criteria.
110
The effectiveness of our internal control over financial reporting as of December 31, 2021 has been audited by EisnerAmper LLP, an independent registered public accounting firm, as stated in their attestation report, which is included herein.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting, except as noted below.
On November 19, 2021, we acquired all the issued and outstanding equity interests of Novitium Pharma LLC (“Novitium”). In conjunction with the transaction, we are currently in the process of integrating Novitium’s policies, processes, people, technology, and operations into the consolidated company, and integrating Novitium’s operations into our system of internal control over financial reporting. As permitted by the Securities and Exchange Commission rules, we excluded Novitium from the assessment of internal control over financial reporting for the year ending December 31, 2021.
Item 9B. Other Information
None.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers, and Corporate Governance
The text of our Code of Ethics, which applies to our principal executive officer, principal financial officer, principal accounting officer or controller, and persons performing similar functions, is posted on our website, www.anipharmaceuticals.com, under the “Governance” subsection of the “Investors” section of the site. We will disclose on our website amendments to, and, if any are granted, waivers of, our Code of Ethics for our principal executive officer, principal financial officer, or principal accounting officer, controller, or persons performing similar functions.
Information required by this item with respect to our directors will be set forth under the caption “Election of Directors” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Information required by this item with respect to our executive officers will be set forth under the caption “Executive Officers of the Company” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Information required by this item with respect to compliance with Section 16(a) of the Exchange Act will be set forth under the caption “Delinquent Section 16(a) Reports” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Information required by this item with respect to our audit committee, our audit committee financial expert, and any material changes to the way in which our security holders may recommend nominees to our Board of Directors will be set forth under the caption “Corporate Governance” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
111
Item 11. Executive Compensation
Information required by this item with respect to executive compensation will be set forth under the caption “Executive Compensation” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information required by this item with respect to security ownership of certain beneficial owners and management will be set forth under the captions “Security Ownership of Certain Beneficial Owners” and “Security Ownership of Directors and Executive Officers” and information relating to our equity compensation plans will be set forth under “Equity Compensation Plan Information” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information required by this item with respect to certain relationships and related transactions and director independence will be set forth under the captions “Certain Relationships and Related Transactions” and “Corporate Governance” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services
Our independent registered public accounting firm is
Information required by this item with respect to principal accounting fees and services will be set forth under the caption “Ratification of Selection of Independent Registered Public Accountants” in our definitive proxy statement for our 2022 annual meeting, to be filed with the SEC pursuant to Regulation 14A no later than 120 days after the close of our fiscal year, and is incorporated herein by reference.
PART IV.
Item 15. Exhibits and Financial Statement Schedules
Documents filed as part of this report on Form 10-K:
(a) Financial Statements:
The consolidated balance sheets of the Registrant as of December 31, 2021 and 2020, the related consolidated statements of operations, statements of comprehensive income, changes in stockholders’ equity, and cash flows for each of the years ended December 31, 2021, 2020, and 2019, the footnotes thereto, and the reports of EisnerAmper LLP, independent registered public accounting firm, are filed herewith.
(b) Financial Statement Schedules:
All schedules have been omitted because they are not applicable or the required information is included in the consolidated financial statements or notes thereto.
(c) Exhibits
Exhibits included or incorporated by reference herein: see Exhibit Index on page 112.
112
ANI PHARMACEUTICALS, INC.
EXHIBIT INDEX TO ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2021
Exhibit |
| Exhibit |
| Method of Filing |
2.1 |
|
| Incorporated by reference to Exhibit 2.1 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on April 12, 2013 (File No. 001-31812) | |
|
|
|
|
|
2.2 |
|
| Incorporated by reference to Exhibit 2.2 to ANI’s Annual Report on Form 10-K as filed for the fiscal year ended December 31, 2013 (File No. 001-31812) | |
2.3 | Incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on March 9, 2021 (File No. 001-31812) | |||
|
|
|
|
|
3.1 |
|
| Incorporated by reference to Exhibit 3.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended June 30, 2013 (File No. 001-31812) | |
|
|
|
|
|
3.2 |
|
| Incorporated by reference to Exhibit 3.1 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on February 16, 2017 (File No. 001-31812) | |
3.3 | Incorporated by reference to Exhibit 3.1 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on November 26, 2021 (File No. 001-31812) | |||
4.1 | Description of Securities | Incorporated by reference to Exhibit 4.1 to ANI’s Annual Report on Form 10-K for the fiscal year ended December 31, 2021 (File No. 001-31812) | ||
4.2 | Registration Rights Schedule to the Merger Agreement, effective as of November 19, 2021 | Incorporated by reference to Exhibit 4.1 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on November 26, 2021 (File No. 001-31812) |
113
Exhibit |
|
Exhibit |
|
Method of Filing |
10.1 |
|
| Incorporated by reference to Exhibit 10.59 to ANI’s Registration Statement on Form S-4 as filed with the Securities and Exchange Commission on December 11, 2012 (File No. 333-185391) | |
|
|
|
|
|
10.2* |
| Employment Agreement, entered into by the Company and James G. Marken |
| Incorporated by reference to Exhibit 10.4 to ANI’s Current Report on Form 8-K filed January 22, 2020 (File No. 001-31812) |
10.3 |
|
| Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2015 (File No. 001-31812) | |
|
|
|
|
|
10.4 |
|
| Incorporated by reference to Exhibit 10.2 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2015 (File No. 001-31812) | |
|
|
|
|
|
10.5* |
|
| Incorporated by reference to Appendix A to the Registrant’s Definitive Proxy Statement on Schedule 14A filed with the Commission on April 14, 2016 | |
|
|
|
|
|
10.6 |
| Asset Purchase Agreement between H2-Pharma, LLC and ANI Pharmaceuticals, Inc. (2) |
| Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2016 (File No. 001-31812) |
|
|
|
|
|
10.7 |
| Asset Purchase Agreement between Cranford Pharmaceuticals, LLC and ANI Pharmaceuticals, Inc. (2) |
| Incorporated by reference to Exhibit 10.2 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2016 (File No. 001-31812) |
|
|
|
|
|
10.8* |
| Employment Agreement, entered into by the Company and Stephen P. Carey |
| Incorporated by reference to Exhibit 10.2 to ANI’s Current Report on Form 8-K filed January 22, 2020 (File No. 001-31812) |
|
|
|
|
|
10.9 |
| Asset Purchase Agreement between Cranford Pharmaceuticals, LLC and ANI Pharmaceuticals, Inc. (2) |
| Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017 (File No. 001-31812) |
114
Exhibit | Exhibit | Method of Filing | ||
10.10 |
| Asset Purchase Agreement between Holmdel Pharmaceuticals, LP and ANI Pharmaceuticals, Inc. (2) |
| Incorporated by reference to Exhibit 10.2 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017 (File No. 001-31812) |
|
|
|
|
|
10.11 |
|
| Incorporated by reference to Exhibit 10.25 to ANI’s Annual Report on Form 10-K for the fiscal year ended December 31, 2017 (File No. 001-31812) | |
|
|
|
|
|
10.12 |
|
| Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2018 (File No. 001-31812) | |
10.13 |
| Amended and Restated Credit Agreement between Citizens Bank, N.A. and ANI Pharmaceuticals, Inc. (2) |
| Incorporated by reference to Exhibit 10.22 to ANI’s Annual Report on Form 10-K for the fiscal year ended December 31, 2018 (File No. 001-31812) |
10.14 |
|
| Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2019 (File No. 001-31812) | |
10.15 |
| Asset Purchase Agreement between Amerigen Pharmaceuticals LTD. and ANI Pharmaceuticals, Inc. |
| Incorporated by reference to Exhibit 10.24 to ANI’s Annual Report on Form 10-K for the fiscal year ended December 31, 2019 (File No. 001-31812) |
10.16 | ANI Pharmaceuticals, Inc. Sixth Amended and Restated 2008 Incentive Plan | Incorporated by reference to Exhibit 10.1 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended June 30, 2020 (File No. 001-31812) | ||
10.17* |
|
| Incorporated by reference to Appendix A to ANI’s Definitive Proxy Statement for the 2020 Virtual Annual Meeting filed on April 23, 2020 (File No. 001-31812) | |
10.18* |
|
| Incorporated by reference to Exhibit 10.20 to ANI’s Annual Report on Form 10-K for the fiscal year ended December 31, 2020 (File No. 001-31812) | |
10.19* | Employment Agreement between Nikhil Lalwani and ANI Pharmaceuticals, Inc., dated July 24, 2020 | Incorporated by reference to Exhibit 10.1 to ANI’s Current Report on Form 8-K filed August 3, 2020 (File No. 001-31812) | ||
10.20* | Incorporated by reference to Exhibit 10.2 to ANI’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2020 (File No. 001-31812) | |||
10.21 | Incorporated by reference to Exhibit 10.1 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on November 26, 2021 (File No. 001-31812) |
115
Exhibit |
| Exhibit |
| Method of Filing | |||
10.22 | Incorporated by reference to Exhibit 10.2 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on March 9, 2021 (File No. 001-31812) | ||||||
10.23* | Incorporated by reference to Exhibit 10.3 to ANI’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on November 26, 2021 (File No. 001-31812) | ||||||
10.24 | Sublicense Agreement, dated as of October 30, 2009, by and between ANIP Acquisition Company, d/b/a ANI Pharmaceuticals, Inc., and Jazz Pharmaceuticals, Inc. (2) | Filed herewith | |||||
10.25 | Master Product Development and Collaboration Agreement, dated as of July 11, 2011, by and among ANIP Acquisition Company d/b/a ANI Pharmaceuticals, Inc. and RiconPharma LLC (2) | Filed herewith | |||||
10.26* | Employment Agreement between and Christopher Mutz and the Company, dated February 10, 2021. | Filed herewith | |||||
10.27* | Employment Agreement between Ori Gutwerg and the Company, dated January 18, 2021. | Filed herewith | |||||
10.28* | Employment Agreement between Chad Gassert and the Company, dated March 8, 2021. | Filed herewith | |||||
21 | Filed herewith | ||||||
23.1 | Filed herewith | ||||||
31.1 |
| Certification of Chief Executive Officer Pursuant to SEC Rule 13a-14 |
| Filed herewith | |||
|
|
|
|
| |||
31.2 |
| Certification of Chief Financial Officer Pursuant to SEC Rule 13a-14 |
| Filed herewith | |||
32.1 |
|
| Furnished herewith | ||||
101 |
| The following financial information from this annual report on Form 10-K for the fiscal year ended December 31, 2021, formatted in Inline XBRL: (i) the audited consolidated Balance Sheets, (ii) the audited consolidated Statements of Operations, (iii) the audited consolidated Statements of Comprehensive Income, (iv) the audited consolidated Statements of Mezzanine Equity and Stockholders’ Equity; (v) the audited consolidated Statements of Cash Flows, and (vi) Notes to consolidated Financial Statements. |
| Filed herewith | |||
116
Exhibit |
| Exhibit |
| Method of Filing | |||
104 | The cover page from the Company Annual Report on Form 10-K for the year ended December 31, 2021 formatted in inline XBRL (included in Exhibit 101) | Filed herewith | |||||
(1) All exhibits to this exhibit have been omitted pursuant to Item 601(b)(2) of Regulation S-K. ANI will furnish the omitted exhibits to the SEC upon request by the SEC.
(2) Confidential treatment has been granted with respect to redacted portions of this document or certain information has been omitted from this exhibit in accordance with Regulation S-K Item 601(b)(10)(iv). The Company agrees to furnish supplementally a copy of any omitted information to the Securities and Exchange Commission upon its request.
* Management contract or compensatory plan or arrangement required to be filed as an exhibit to this Annual Report on Form 10-K pursuant to Item 15(a).
117
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ANI PHARMACEUTICALS, INC. |
| |
|
|
|
|
|
|
By: | /s/ Nikhil Lalwani |
|
| Nikhil Lalwani |
|
| President and Chief Executive Officer |
|
| (principal executive officer) |
|
|
|
|
Date: March 15, 2022 | ||
By: | /s/ Stephen P. Carey |
|
| Stephen P. Carey |
|
| Senior Vice President, Finance and |
|
| Chief Financial Officer |
|
| (principal financial and accounting officer) |
|
|
|
|
Date: March 15, 2022 |
| |
Pursuant to the requirements the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Name |
| Capacity |
| Date |
|
|
|
|
|
/s/ Nikhil Lalwani |
| Director, President, and |
| March 15, 2022 |
Nikhil Lalwani |
| Chief Executive Officer (principal executive officer) |
|
|
|
|
|
|
|
/s/ Stephen P. Carey Stephen P. Carey | Senior Vice President, Finance and Chief Financial Officer (principal financial and accounting officer) | March 15, 2022 | ||
/s/ Muthusamy Shanmugam | Director, Head of Research and Development and Chief Operating Officer of New Jersey Operations | March 15, 2022 | ||
Muthusamy Shanmugam | ||||
/s/ Patrick D. Walsh |
| Director and Chairman of the Board of |
| March 15, 2022 |
Patrick D. Walsh |
| Directors |
|
|
|
|
|
|
|
/s/ Thomas J. Haughey |
| Director |
| March 15, 2022 |
Thomas J. Haughey |
|
|
|
|
|
|
|
|
|
/s/ David B. Nash, M.D., M.B.A. |
| Director |
| March 15, 2022 |
David B. Nash, M.D., M.B.A. |
|
|
|
|
|
|
|
|
|
/s/ Robert E. Brown, Jr. |
| Director |
| March 15, 2022 |
Robert E. Brown, Jr. |
|
|
|
|
|
|
|
|
|
/s/ Jeanne Thoma |
| Director |
| March 15, 2022 |
Jeanne Thoma | ||||
/s/ Antonio Pera | Director | March 15, 2022 | ||
Antonio Pera | ||||
119
Exhibit 10.24
Certain information has been excluded from this agreement (indicated by “[***]”) because such information is both not material and the type that the registrant treats as private or confidential.
SUBLICENSE AGREEMENT
This Sublicense Agreement (the “Agreement”) is entered into as of the 30th day of October, 2009 by and between ANIP ACQUISITION COMPANY, d/b/a ANI PHARMACEUTICALS, INC., a Delaware corporation (“ANI”), and JAZZ PHARMACEUTICALS, INC., a Delaware corporation (“Jazz Pharmaceuticals”).
WHEREAS, ANI and Jazz Pharmaceuticals have entered into a Manufacturing and Supply Agreement (the “Supply Agreement”), dated as of January 25, 2008, whereby ANI has agreed to manufacture and supply Jazz Pharmaceuticals requirements for Luvox®-IR (fluvoxamine maleate) (the “Branded Product”);
WHEREAS, in connection with the Supply Agreement, ANI wishes to acquire the right to manufacture and market an unbranded generic version of the Branded Product under ANI’s label (the “Generic Product”); and
WHEREAS, Jazz Pharmaceuticals acquired the rights to the Branded Product pursuant to that certain License Agreement (the “Solvay License Agreement”), dated as of January 31, 2007 by and between Jazz Pharmaceuticals and Solvay Pharmaceuticals, Inc. (“Solvay”), and Jazz Pharmaceuticals wishes to sublicense ANI the right to manufacture, package, use and sell (and have sold) the Generic Product on the terms and conditions set forth in this Agreement.
NOW, THEREFORE, in consideration of the mutual covenants and agreements set forth in this Agreement, and for other good and valuable consideration, the receipt of which is hereby acknowledged, the parties hereto agree as follows:
1. Grant of Sublicense.
1.1 Sublicense. Jazz Pharmaceuticals hereby grants to ANI a sublicense, without the right to further sublicense, to make, package, use and sell (and have sold) the Generic Product in the United States (the “License”). The rights granted to ANI hereunder, including the NDA Transfer (as defined below) are granted subject to the rights of Solvay pursuant to the Solvay License Agreement. Jazz Pharmaceuticals hereby represents and warrants to ANI that (a) the execution, delivery and performance of this Agreement by Jazz Pharmaceuticals does not conflict with or constitute a breach of any order, judgment, agreement, or instrument to which it is a party, including, without limitation, the Solvay License Agreement; and (b) the execution, delivery and performance of this Agreement by Jazz Pharmaceuticals does not require the consent of any person, which consent has not been obtained.
1.2 Trademarks. ANI agrees and acknowledges that it shall not acquire by virtue of this Agreement any interest in or to any trademarks or trade names of Jazz Pharmaceuticals or Solvay, including, but not limited to, the brand name Luvox®.
1.3 Pharmacovigilance Agreement. ANI and Jazz Pharmaceuticals hereby agree that effective as of the date hereof, the Pharmacovigilance Agreement entered into between the parties on August 22, 2008 is terminated and of no further force or effect.
1.4 Supply Agreement. The parties agree to enter into a supply agreement for supply of fluvoxamine maleate (“API”) in the form attached hereto as Exhibit A (the “API Supply Agreement”), pursuant to which Jazz Pharmaceuticals have manufactured ANI’s requests of API for ANI during the term thereof.

2. NDA Transfer.
2.1 Transfer. Jazz Pharmaceuticals agrees to transfer responsibility for the New Drug Application #21-519 (the “NDA”) to ANI (the “NDA Transfer”) as soon as is reasonably practicable after entering into this Agreement including by so notifying the FDA of such NDA Transfer. ANI agrees to promptly notify the FDA of the NDA Transfer.
2.2 Right of Reference. Immediately upon the NDA Transfer, ANI grants to Jazz Pharmaceuticals the right to reference the NDA for all purposes.
2.3 Ongoing Requirements. ANI agrees to comply with and to provide documentation evidencing compliance with its ongoing reporting and maintenance requirements to the FDA related to the NDA. Such evidence of compliance may consist of a copy of any Form 356-H filed with the FDA when meeting the obligations in the first sentence of this Section 2.3. ANI agrees to provide a copy of any notice from the FDA indicating an adverse finding by the FDA related to the NDA (the “Adverse Finding Letter”).
2.4 NDA Reversion. In the event ANI receives an Adverse Finding Letter from the FDA relating to the NDA and ANI is either not able to cure or provide evidence of a reasonable plan to cure any issues raised by the FDA in an Adverse Finding Letter within 30 days of receipt by ANI of such Adverse Finding Letter, Jazz Pharmaceuticals may, at its sole discretion, request in writing (the “Reverse NDA Transfer Notice”) that ANI transfer responsibility for the NDA to Jazz Pharmaceuticals (the “Reverse NDA Transfer”). ANI agrees that upon receipt of a Reverse NDA Transfer Notice that it will immediately perform any actions necessary to accomplish the Reverse NDA Transfer, including the sending of any notices to the FDA.
3. Compensation.
3.1 Royalty Payments. As consideration for the sublicense granted by Jazz Pharmaceuticals to ANI hereunder, ANI shall pay to Jazz Pharmaceuticals royalty payments in each calendar year during the term of the Agreement equal to [***] of the Generic Product’s Net Sales (defined below). For purposes of this Agreement, “Net Sales” shall mean the gross amounts invoiced by ANI, its affiliates and sublicensees on all sales of the Generic Product to independent unrelated third parties in bona fide arms’ length transactions, less (a) transportation, shipping and freight charges, including insurance and handling, to the extent that such charges are included in the gross amounts invoiced in connection with the transport of the Generic Product; (b) sales, use and excise taxes, value added taxes, and duties which fall due and are paid as a consequence of such sales by ANI or its affiliates or sublicensees and any other governmental charges imposed upon the importation, use or sale of the Generic Product; and (c) the following deductions actually allowed and taken by such third parties and not otherwise recovered by or reimbursed to ANI or its affiliates and sublicensees: (i) trade, quantity and cash discounts; (ii) allowances or credits on account of rejection, defects, recall or return of the Generic Product or on account of retroactive price reductions or wholesaler chargebacks affecting such Generic Product; and (iii) rebates, refunds, reductions and chargebacks specifically related to the Generic Product including those granted to insurers, buying groups, government agencies or similar bodies.
3.2 Records. ANI shall keep complete and accurate records of all sales of the Generic Product and the calculation of Net Sales of the Generic Product. Jazz Pharmaceuticals shall have the right, at Jazz Pharmaceuticals’ expense and after thirty (30) days’ prior written notice to ANI, through an independent certified public accountant, on a mutually agreeable date, to examine such records at any time within two (2) years after the due date of the royalty payments to which such records relate (but no more than once each calendar year) during regular business hours, during the term of this Agreement and for twelve (12) months after its expiration or termination, in order to verify the accuracy of the reports to be made under Section 3.3 hereunder. The results of such examination will be made available to ANI. If, thereafter, ANI disputes in good faith the accuracy of the results of such examination, the parties will retain a second independent certified public accountant whose examination will be binding upon both parties. The parties will share the expense of such examination equally.
3.3 Reports. Within forty-five (45) days after the end of each calendar quarter during the term of this Agreement, ANI shall provide Jazz Pharmaceuticals with a written report of estimated Net Sales of the Generic Product during such quarter. Within ninety (90) days after the end of each calendar year, a True-Up of Net Sales will be performed. Simultaneously with the submission of such quarterly report, ANI shall pay to Jazz Pharmaceuticals all royalty payments due to Jazz Pharmaceuticals under Section 3.1 hereof. Interest, at a rate of twelve percent (12%) per annum, or at the highest legal rate if less than 12%, shall be payable for any late payments.
3.4 Payment Mechanics, Taxes. All payments will be made by wire transfer to an account designated by Jazz Pharmaceuticals to ANI in writing. All undisputed payments not made when due hereunder will bear interest at the rate stated in Section 3.3 from the date the payment became due. ANI shall be responsible for the payment of, and shall promptly pay, all federal, state, and local transfer, sales, and other taxes, if any, levied or imposed on Jazz and ANI as a result of the transactions contemplated by this Agreement, including without limitation sales and use taxes but excluding any tax payable on any income or gain of Jazz Pharmaceuticals.
4. Effective Date and Term.
4.1 Effective Date and Term. This Agreement is effective on and as of the Effective Date and, unless terminated in accordance with any of the provisions hereof, will remain in full force and effect thereafter.
5. Indemnification.
5.1 Indemnity. ANI will indemnify, defend and hold Jazz Pharmaceuticals, its affiliates, successors, permitted assigns and their respective officers, directors, managers, members, stockholders, partners and employees harmless from and against any and all liabilities, claims, demands, damages, costs, expenses or money judgments incurred by or rendered against ANI which arise out of the use, labeling or manufacture, processing, packaging, sale or commercialization of the Generic Product by ANI, its subcontractors, distributors, and marketing partners. Jazz Pharmaceuticals will permit ANI’s attorneys, at ANI’s discretion and cost, to control the defense of any claims or suits as to which Jazz Pharmaceuticals may be entitled to indemnification hereunder, and Jazz Pharmaceuticals agrees not to settle any such claims or suits without the prior written consent of ANI. Jazz Pharmaceuticals will have the right to participate, at its own expense, in the defense of any such claim or demand to the extent it so desires.
5.2 Notice. Jazz Pharmaceuticals will give ANI prompt notice in writing, in the manner set forth in Section 8.6 below, of any claim or demand made against Jazz Pharmaceuticals for which Jazz Pharmaceuticals may be entitled to indemnification under Section 5.1.
6. Disclaimers.
JAZZ PHARMACEUTICALS DISCLAIMS ANY EXPRESS OR IMPLIED WARRANTY (A) THAT THE GENERIC PRODUCT, OR THE MANUFACTURE, USE OR SALE THEREOF, WILL BE FREE FROM CLAIMS OF PATENT INFRINGEMENT, INTERFERENCE OR UNLAWFUL USE-OF
PROPRIETARY INFORMATION OF ANY THIRD PARTY AND (B) OF THE ACCURACY, RELIABILITY, TECHNOLOGICAL OR COMMERCIAL VALUE, COMPREHENSIVENESS OR MERCHANTABILITY OF THE PRODUCT OR ANY TECHNOLOGY INCORPORATED THEREIN OR ITS SUITABILITY OR FITNESS FOR ANY PURPOSE WHATSOEVER. JAZZ PHARMACEUTICALS DISCLAIMS ALL OTHER WARRANTIES OF WHATEVER NATURE, EXPRESS OR IMPLIED.
7. Termination.
7.1 Termination by Jazz Pharmaceuticals. Jazz Pharmaceuticals may, in its discretion, terminate this Agreement:
(a) if the Solvay License Agreement is terminated in accordance with the terms set forth therein;
(b) if ANI breaches or defaults in the performance or observance of any material provisions of this Agreement, or the API Supply Agreement and such breach or default is not cured within sixty (60) days after written notice by Jazz Pharmaceuticals specifying such breach or default (or if such breach or default is not of a type which can reasonably be cured in sixty (60) days, then such longer period as is reasonable);
(c) if ANI enters into any proceeding, whether voluntary or otherwise, in bankruptcy, reorganization or arrangement for the appointment of a receiver or trustee to take possession of ANI’s assets or any other proceedings under any law for the relief of creditors or makes an assignment for the benefit of its creditors;
(d) if Jazz Pharmaceuticals delivers, pursuant to the terms of Section 2.4, a Reverse NDA Transfer Notice to ANI;
(e) if Jazz Pharmaceuticals terminates the Supply Agreement pursuant to the terms of the Supply Agreement; or
(f) if ANI does not make the royalty payments when due pursuant to Section 3.1.
7.2 Termination by ANI. ANI may terminate this Agreement with the consent of Jazz Pharmaceuticals, such consent not to be unreasonably withheld.
7.3 Consequences of Termination. Termination of this Agreement for any reason in accordance with the terms hereof will be without prejudice to any remedies which either party may then or thereafter have hereunder or otherwise; If this Agreement terminates pursuant to this Section 7, ANI will immediately discontinue any promotion and sales of the Generic Product, if so requested by Jazz Pharmaceuticals.
8. Miscellaneous.
8.1 Waiver; Remedies and Amendment. Any waiver by any party hereto of a breach of any provisions of this Agreement will not be implied and will not be valid unless such waiver is recited in writing and signed by such party. Failure of any party to require, in one or more instances, performance by the other party or parties in strict accordance with the terms and conditions of this Agreement will not be deemed a waiver or relinquishment of the future performance of any such terms or conditions or of any other terms and conditions of this Agreement. A waiver by any party of any term or condition of this Agreement, including this Section 8.1, shall be valid only if in writing and will not be deemed or construed to be a waiver of such term or condition for any other term. All rights, remedies, undertakings, obligations and agreements contained in this Agreement will be cumulative and none of them will be a limitation of any other remedy, right, undertaking, obligation or agreement of any party. This Agreement may not be amended except in a writing signed by all parties.
8.2 Assignment, No Inconsistent Agreements. ANI may not assign its rights and obligations hereunder without the prior written consent of Jazz Pharmaceuticals. Neither Jazz Pharmaceuticals nor ANI will enter into any agreement that is inconsistent with its obligations hereunder. Upon written notice to ANI, Jazz Pharmaceuticals may assign its rights and obligations hereunder and under the Solvay License Agreement, without the consent of ANI.
8.3 Counterparts. This Agreement may be executed in any number of counterparts, each of which when so executed will be deemed to be an original and all of which when taken together will constitute this Agreement.
8.4 Governing Law. This Agreement will be governed by and construed in accordance with the laws of the state of New York as applied to residents of that state entering into contracts to be performed in that state.
8.5 Headings. The headings set forth at the beginning of the various sections of this Agreement are for convenience and form no part of the Agreement between the parties.
8.6 Notices. All notices, requests, instructions, consents and other communications to be given pursuant to this Agreement shall be in writing and shall be deemed received (a) on the same day if delivered in person, by same-day courier or by telegraph, telex, facsimile, electronic mail or other electronic transmission, (b) on the next day if delivered by overnight mail or courier, or (c) on the date indicated on the return receipt, or if there is no such receipt, on the third calendar day (excluding Sundays) if delivered by certified or registered mail, postage prepaid, to the party for whom intended to the following addresses:
If to Jazz Pharmaceuticals:
Jazz Pharmaceuticals, Inc.
3180 Porter Drive Palo Alto,
CA 94304
Attn: General Counsel
If to ANI:
ANIP Acquisition Company
d/b/a ANI Pharmaceuticals Inc.
210 Main Street West
Baudette, MN 56623
Attention: President & CEO
With a copy to:
Sonnenschein Nath & Rosenthal LLP
1221 Avenue of the Americas, 25th Floor
New York, NY 10020
Attn: Ms. Jane A. Meyer
Each party may by written notice given to the other in accordance with this Agreement change the address to which notices to such party are to be delivered.
8.7 Severability. If any provision of this Agreement is held by a court of competent jurisdiction to be invalid or unenforceable, it will be modified, if possible, to the minimum extent necessary to make it valid and enforceable or, if such modification is not possible, it will be stricken and the remaining provisions will remain in full force and effect.
8.8 Survival. The provisions of Sections 3.2, 3.3, 3.4, 5, 6, 8.1, 8.2, 8.4, 8.5, 8.6, 8.7 and this Section 8.8 will survive the termination for any reason of this Agreement.
8.9 Force Majeure. No party to this Agreement will be liable for failure or delay in the performance of any of its obligations hereunder, if such failure or delay is due to causes beyond its reasonable control including, without limitation, acts of God, earthquakes, fires, strikes, acts of war, or intervention of any governmental authority, but any such delay or failure will be remedied by such party as soon as possible after the removal of the cause of such failure or delay.
IN WITNESS WHEREOF, the parties have executed this Agreement on the date first set forth above. | |||
ANIP ACQUISITION COMPANY d/b/a ANI PHARMACEUTICALS, INC. | |||
By: | /s/ Charlotte C. Arnold | ||
Name: | Charlotte C. Arnold | ||
Title: | Chief Financial Officer | ||
JAZZ PHARMACEUTICALS, INC. | |||
By: | /s/ Bob Myers | ||
Name: | Bob Myers | ||
Title: | President | ||
Exhibit A
API Supply Agreement
[AGREEMENT EXPIRED]
Exhibit 10.25
Certain information has been excluded from this agreement (indicated by “[***]”) because such information is both not material and the type that the registrant treats as private or confidential.
MASTER PRODUCT DEVELOPMENT AND COLLABORATION AGREEMENT
BY AND AMONG
RICONPHARMA LLC
AND
ANIP ACQUISITION COMPANY d/b/a ANI PHARMACEUTICALS, INC.
JULY 2011
This MASTER PRODUCT DEVELOPMENT AND COLLABORATION AGREEMENT is made as of the 11th day of July 2011, by and among ANIP Acquisition Company d/b/a ANI Pharmaceuticals, Inc., a corporation organized and existing under the laws of the State of Delaware (“ANI”), having a place of business at 210 Main Street West, Baudette, MN 56623 and RiconPharma LLC, a limited liability company having its principal office at 100 Ford Road, Suite #9, Denville, NJ 07834 (“RiconPharma”). ANI and RiconPharma may each be referred to herein individually as a “Party” and collectively as the “Parties.”
WHEREAS, RiconPharma possesses expertise relating to the development of finished dosage forms of pharmaceutical products and ANI possesses expertise relating to the manufacture of finished pharmaceutical products and also possesses expertise relating to the marketing, distribution and sale of pharmaceutical products;
WHEREAS, the Parties desire, from time to time, to collaborate in a cost, asset, and profit sharing arrangement for the development, manufacturing, regulatory approval, and marketing of pharmaceutical products in the US and also to invest their respective resources in developing, obtaining regulatory approval for manufacturing and marketing such products in the manner to be set forth in a Amending Product Exhibit hereto (each, a “Amending Product Exhibit”); and
WHEREAS, each Amending Product Exhibit shall delineate the specific terms and conditions related to each new Product collaboration including, but not limited to, a description of the Product (hereinafter defined) to be developed, the estimated cost of development, and the percentage allocation of costs and profits; and
NOW, THEREFORE, in consideration of the foregoing and of the agreements, representations, covenants, and warranties contained herein and other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the Parties hereby agree as follows:
1. DEFINITIONS.
For the purposes of this Agreement, the following terms shall have the following meanings:
1.1 “Affiliate” shall mean any person, directly or indirectly, controlling, controlled by, or under common control with, another person. Without limiting the generality of the foregoing, a person is considered to be in control of or to be controlled by another person if such person holds 50% or more of the outstanding voting equity interest in such other person or such other person holds 50% or more of its outstanding voting equity interest.

1.2 “ANDA” shall mean an abbreviated new drug application or similar health registration application that is or will be filed with a Regulatory Authority to obtain Regulatory Approval to market a Product in the Territory.
1.3 “API” shall mean an active pharmaceutical ingredient in a Product shipped to ANI.
1.4 “API Suppliers” shall have the meaning set forth in Section 7.2 of this Agreement.
1.5 “Approved Product” shall mean a Product that shall have been granted all necessary approvals by all required Regulatory Authorities sufficient to permit the marketing and sale by ANI or an Affiliate of ANI of such Product in the Territory.
1.6 “Base Price” shall mean the price charged by ANI for a Unit of Product, which price shall be comprised of the cost of Raw Materials, Components, labor, overhead and profit per Unit, which is further described in the Amending Exhibit.
1.7 “Batch” with respect to a Product, shall mean a specific quantity of a Product that is intended to have uniform character and quality within specified limits, and is produced according to a single manufacturing order during the same cycle of manufacture, and designated by a batch number.
1.8 “Bioequivalent Product” shall mean with respect to a Product, a drug product that is a bioequivalent, as that term is used in the FDA Orange Book and that is identical in strength or concentration, contains the same active ingredient(s), is in the same dosage form, and utilizes the same route of administration as the Product.
1.9 “Bioequivalence Study” shall have the meaning set forth in Section 3.1 of this Agreement.
1.10 “Calendar Quarter” shall mean the periods of three consecutive calendar months ending on March 31, June 30, September 30 or December 31.
1.11 “Certificate of Analysis” shall mean a document, which is dated and signed by a duly authorized representative of the quality control or quality assurance department of ANI, certifying that a Batch of any Product meets all Specifications accompanied by the certificate(s) of analysis prepared and signed by any manufacturer(s) of the Product(s) in the Batch and Raw Materials for the Product in the Batch certifying that the Products and Raw Materials meet all applicable specifications.
1.12 “Claim” shall have the meaning set forth in Section 17.5 of this Agreement.
1.13 “Commercially Reasonable Efforts” with respect to the efforts to be expended by a Party regarding any objective under this Agreement, shall mean reasonable, diligent, good-faith efforts to accomplish such objective as a reasonable person or entity similarly situated would normally use to accomplish a similar objective under similar circumstances exercising reasonable business judgment.
1.14 “Components” shall mean all labels, bottles, caps, seals, cardboard packaging, inserts and other materials (excluding Raw Materials) used to Label and package a Unit for shipment to ANI.
1.15 “Confidential Information” shall have the meaning set forth in Section 21 of this Agreement.
1.16 “Continuing Party” or “Continuing Parties” shall have the meaning set forth in Section 20.6 of this Agreement.
1.17 “Cost of Goods” shall mean the total amount contingently charged by ANI for the purchase of Products during any Calendar Quarter, each Unit invoiced at the Base Price.
1.18 “Development Costs” shall mean the costs incurred by the Parties in developing a Product, including but not limited to costs of formulation development, analytical development, scale-up, demo batches, bio-batch manufacturing, ICH Stability packaging and testing, bioequivalence studies, and ANDA filings.
1.19 “Development Cost Percentage” shall mean the percentage of Development Costs allocated to each Party in connection with developing Products and as set forth in the Amending Product Exhibit.
1.20 “DMF” shall mean the drug master file, confidential or otherwise, covering the manufacture and analysis of an API with respect to a Product in the Territory.
1.21 “EDC” shall have the meaning set forth in Section 3.2.
1.22 “Effective Date” shall mean the date of execution by the Parties of the first Amending Product Exhibit to this Agreement.
1.23 “FDA” shall mean the United States Food and Drug Administration, or any successor agency.
1.24 “FD&C Act” shall mean the Federal Food, Drug and Cosmetic Act of 1938, as amended, and the regulations thereunder, as the same may be amended from time to time.
1.25 “Good Clinical Practices” or “GCPs” shall mean the then-current standards for clinical trials for pharmaceuticals as set forth in the FD&C Act and applicable regulations promulgated thereunder.
1.26 “Good Laboratory Practices” or “GLPs” shall mean the then-current standards for laboratory activities for pharmaceuticals as set forth in the FD&C Act and applicable regulations promulgated thereunder.
1.27 “Good Manufacturing Practices” or “GMPs” shall mean the then-current standards for the manufacture of pharmaceutical Products as set forth in the FD&C Act and applicable regulations promulgated thereunder.
1.28 “Indemnity Claim” shall have the meaning set forth in Section 17.5 of this Agreement.
1.29 “Initial Marketing Date” shall be the date listed on the FDA Form 2657 (New Product Listing Form) or its successor, indicating the first date a Product is distributed to customers in the Territory.
1.30 “Invention(s)” shall mean an invention conceived and reduced to practice in the course of the performance of and within the scope of this Agreement.
1.31 “Know-How” shall mean all proprietary technical and clinical information, data and know-how relating to a Product, whether or not patentable, which is owned or controlled as of the Effective Date or acquired or developed during the term of this Agreement by a Party hereto. Know-How shall include, without limitation, processes, formulas, discoveries and inventions whether relating to biological, chemical, pharmacological, toxicological, pharmaceutical, physical and analytical safety, quality control and clinical data. The term “Know-How” shall exclude: (i) processes, information and data which is, as of the Effective Date, generally available to the public or later becomes generally available without breach by a Party of its obligations of confidentiality hereunder; or (ii) any general development or manufacturing know-how not specific to a Product.
1.32 “Label” and “Labeling” have the meaning given those terms by 21 CFR Part 201.
1.33 “Litigation Expenses” shall mean expenses incurred in investigating, defending, or litigating any claims, demands, or actions related to a Product made or brought by a Third Party (including reasonable legal fees and the payment of damages and expenses to a Third Party).
1.34 “Manufacturer and Development Technology” shall mean all information, data, intellectual property and Know-How, whether patentable or not, which is owned or controlled by ANI or RiconPharma prior to or during the Term and which is necessary or useful in developing and manufacturing Products, in developing and conducting Bioequivalence Studies for the Products, in preparing and filing Regulatory Approvals for the Products and in maintaining such Regulatory Approvals.
1.35 “Net Profits” shall, with respect to any Product, Net Sales less Base Price in respect of such Product.
1.36 “Net Sales” shall mean the gross amounts invoiced by ANI for any Product sold to third less the sum of chargebacks, cash and quantity discounts, returns, rebates and such other credits granted by ANI or taken by customers with respect to such Product sales. Net Sales shall be determined in accordance with generally accepted accounting principles using the accrual method of accounting, consistent with historical practices of ANI. The sum of chargebacks, cash and quantity discounts, returns, rebates and such other credits will not exceed [ *** ] of gross amounts invoiced without the consent of both ANI and RiconPharma.
1.37 “Non-Continuing Party” or “Non-Continuing Parties” shall have the meaning set forth in Section 20.6 below.
1.38 “Party” shall mean either ANI or RiconPharma and “Parties” shall mean two or more of the following as the context dictates: ANI and RiconPharma.
1.39 “Phase(s)” shall have the meaning set forth in the Amending Product Exhibit of any Products in this Agreement.
1.40 “Policy” shall have the meaning set forth in Section 18.1 of this Agreement.
1.41 “Product(s)” shall mean the Product(s) set forth in each Amending Product Exhibit.
1.42 “Product Action” shall have the meaning set forth in Section 11.2 of this Agreement.
1.43 “Profit Sharing Percentage” shall mean the percentage of Net Profits allocated to each Party with respect to each Product as set forth in each Amending Product Exhibit to this Agreement for that Product.
1.44 “Raw Materials” shall mean the API and inactive ingredients used to manufacture a Product.
1.45 “Recall” shall have the meaning set forth in Section 11.1 of this Agreement.
1.46 “Records” shall have the meaning set forth in Section 19.1 of this Agreement.
1.47 “Reference Product” shall mean the product currently marketed and sold under the pharmaceutical brand identified in the Amending Product Exhibit.
1.48 “Registration”, with respect to a Product, shall mean the meeting of all of the requirements of all applicable Regulatory Authorities necessary to permit the commencement of marketing and selling such Product in the Territory by ANI or an Affiliate of ANI.
1.49 “Regulatory Approval” shall mean the authorizations and approvals of any Regulatory Authority (including, without limitation, approvals of ANDAs and NDAs) required for the commercial manufacture, distribution, marketing, promotion, offer for sale, use, import, export and sale of Product(s) in the Territory.
1.50 “Regulatory Authority” shall mean any and all bodies and organizations regulating the manufacture, importation, marketing, distribution, use and sale of any of the Products in the Territory.
1.51 “Retaining Party” shall have the meaning set forth in Section 19.3 of this Agreement.
1.52 “Sales and Marketing Company” shall have the meaning set forth in Section 2.1 of this Agreement.
1.53 “SIR” shall have the meaning set forth in Section 18.1 of this Agreement.
1.54 “Specifications” shall mean the specifications for each Product as agreed to by the Parties and as approved by the applicable Regulatory Authority, which Specifications may be amended from time to time by written agreement between the Parties and as specifically requested by the applicable Regulatory Authority.
1.55 “Term” shall have the meaning set forth in Section 20.1 of this Agreement.
1.56 “Territory” shall mean the United States of America.
1.57 “Third Party” shall mean any person or entity other than a Party or any of its Affiliates.
1.58 “Trademark” means the trade name and/or trademark used and owned by a Party.
1.59 “Unit” shall have the meaning set forth in the Amending Product Exhibit.
2. THE COLLABORATION.
2.1 The Parties agree to collaborate in the selection of Products and in the development, manufacturing, registration and approval, and marketing of such Products as set forth in more detail in this Agreement and any applicable Amending Product Exhibit. Unless otherwise specified in an Amending Product Exhibit, RiconPharma will be responsible for developing the Products and ANI will be responsible for manufacturing and distribution of the Products in the Territory. The Parties shall be jointly responsible for directing any bioequivalence studies and obtaining Regulatory Approval for such pharmaceutical products, and ANI shall be responsible for maintaining such Regulatory Approvals. ANI or a separate sales and marketing company designated by ANI (a “Sales and Marketing Company”) will be primarily responsible for the marketing, distribution and sale of the Products as well as customer service, rebate management, billing, warehousing and such other responsibilities as are regularly performed by a pharmaceutical distributor.
2.2 The Parties shall jointly own all the rights, title, and interest in the Products (including without limitation, the ANDA for the Products). The respective percentages of ownership for each Product shall be one-half for each Party unless a different percentage is set forth in the Amending Product Exhibit for that Product. Subject to Section 2.3, neither Party, without the prior written consent of the other Party, which consent shall not be unreasonably withheld, conditioned or delayed, may: (i) assign this Agreement; (ii) sell or assign its ownership rights to a Product to a Third Party; or (iii) license or assign any other right, title, or interest in a Product.
2.3 In the event of a sale of more than 50% of a Party’s total assets, as reflected in the Party’s most recent annual financial statements, that Party may assign this Agreement or sell or assign its ownership rights to a Product without the prior written consent of the other Party. It is understood that any successor company,
either through assignment or acquisition of the ownership rights from any Party, shall be bound by the terms and conditions contained within this Agreement.
2.4 ANI or a Sales and Marketing Company designated by ANI shall have the exclusive rights to market, distribute, offer for sale, and sell Products in the Territory during the Term of this Agreement.
2.5 True and complete copies of any Party’s agreements with any Third Party or Affiliate for the manufacture or supply of any Product, Raw Materials or Components shall be attached as exhibits to this Agreement simultaneously with the delivery of an Amending Product Exhibit contemplating the inducement of such Third Party or Affiliate.
2.6 The Parties hereby appoint ANI as an Authorized Distributor of Record (“ADR”) for all Products under this Agreement, and authorize ANI to designate additional ADRs on behalf of ANI for all Products.
3. DEVELOPMENT.
3.1 RiconPharma shall be responsible for developing the Products, including, but not limited to, development of the Products, formulations, analytical methods, and the performance and coordination, including oversight, of any necessary clinical studies with ANI to determine if such Products are Bioequivalent to the Reference Products. Such clinical studies shall be conducted by Third Party contract research organizations selected by the Parties (each, a “Bioequivalence Study”). If a Bioequivalence Study does not demonstrate that the formulations developed by RiconPharma are bioequivalent to the Reference Products, and if the Parties determine that an additional Bioequivalence Study is advisable, then RiconPharma shall reformulate the Products for use in additional Bioequivalence Studies and any actual additional documented costs specifically attributed to the reformulation shall be shared by the Parties according to each Party’s Development Cost Percentage set forth in the Amending Product Exhibit.
3.2 Development Costs for each Phase of a Product will be as set forth in the Amending Product Exhibit for that Product, and shall be shared by the Parties in accordance with each Party’s Development Cost Percentage for that Product. Subject to Section 3.5, if a Party actually incurs Development Costs during a Phase in excess of that Party’s share of Development Costs for the Phase (the “EDC”), the other Party shall, to the extent not already paid, reimburse that Party its respective share of the EDC within thirty (30) days after receipt by the other Party of written notice from the Party requesting reimbursement. Each notice must include the amount requested and the written support for the EDC.
3.3 For each Product, RiconPharma shall use its Commercially Reasonable Efforts to complete each Phase of development by the Completion Date for that Phase indicated in the Amending Product Exhibit for that Product.
3.3.1 Without limitation, RiconPharma will be responsible for all development functions (e.g. pre-formulations, formulations and analytical method development, technology transfer and scale-up support), and will provide necessary support in obtaining Regulatory Approval for such Products, and all such other responsibilities as are typically undertaken by a company engaged in pharmaceutical development.
3.3.2 Without limitation, ANI will be responsible for manufacturing the scale-up and demo batches, ICH stability testing, CMC, Biobatches, ANDA submissions, validations and all such other responsibilities as are typically undertaken by a company engaged in pharmaceutical manufacturing.
3.3.3 Without limitation, ANI or a Sales and Marketing Company designated by ANI will be responsible for sales, marketing and distribution and all such other responsibilities as are typically undertaken by a company engaged in pharmaceutical distribution.
3.4 For each Product, RiconPharma will provide ANI with written notice of the completion of each Phase of development set forth in the Amending Product Exhibit for that Product and ANI will have ten (10) business days after the receipt of such notice within which to notify RiconPharma of any termination of that Amending Product Exhibit as provided in Section 20.4.
3.5 No Party shall have any obligation to reimburse the other Party for any portion of expenses incurred by a Party in excess of 105% of the anticipated Development Costs set forth in an Amending Product Exhibit unless such excess expenses have been approved, in writing, by all Parties prior to the time they are incurred
3.6 In the event that the Parties mutually agree to accept payment from a Third Party in exchange for not developing, manufacturing or marketing a Product, such payment shall be shared by the Parties in accordance with each Party’s Profit Sharing Percentage set forth in the Amending Product Exhibit.
4. COMMERCIALIZATION.
4.1 The Parties may market the Products under a trade name which ANI or a Sales and Marketing Company designated by ANI shall have the right to select, subject to the approval of both Parties, which approval will not be unreasonably withheld, conditioned or delayed. ANI shall own all trade names. The costs of searching, selecting, registering and enforcing a trade name for a Product, if any, shall be advanced by ANI. ANI shall be responsible for responding to regulatory inquiries relating to any trade names selected hereunder.
4.2 All Products sold by ANI shall bear the ANI Trademark and the applicable ANI NDC number and labeler code. To the extent permitted by applicable law and regulations, ANI shall be identified on the Product packaging as the manufacturer of the Products and ANI shall be identified on the Product packaging as the labeler/distributor of the Product.
4.3 ANI agrees that at all times during the Term, neither it nor any of its Affiliates will develop, manufacture, sell or distribute a product that is the same as a Product or a product that is a Bioequivalent Product of a Product in the Territory unless RiconPharma approves in writing.
4.4 RiconPharma agrees that at all times during the Term, neither it nor any of its Affiliates will develop, manufacture, sell or distribute a product that is the same as a Product or a product that is a Bioequivalent Product of a Product in the Territory unless ANI approves in writing.
4.5 All Parties may manufacture, distribute, promote, and sell products in the Territory other than (i) the Products, (ii) Bioequivalent Products of the Products, or (iii) a product that is the same as a Product, and may acquire such products from Third Party manufacturers.
4.6 ANI shall consult with RiconPharma and determine the price at which it shall sell the Products in the Territory. ANI shall have sole discretion over the pricing, marketing, and sales of Products in the Territory including, but not limited to, marketing strategy, sales strategy, Product placement, Product distribution, the terms of sale of Products, and decisions whether to accept returns on Products from the Territory.
4.7 ANI shall at all times maintain a ninety (90) day rotating inventory of API and Components for all Products covered by an Amending Product Exhibit not terminated pursuant to Section 20.
4.8 ANI and RiconPharma shall promptly provide or make available for review all testing documentation, material safety data sheets, certificates of analysis and all similar materials as reasonably requested by its Customers.
5. SHARING OF NET PROFITS.
5.1 Net Profits from the sale of a Product will be shared by the Parties in accordance with each Party’s Profit Sharing Percentage set forth in the Amending Product Exhibit.
5.2 Within thirty (30) days after the end of each Calendar Quarter, ANI shall calculate the Net Sales and the Net Profits obtained from the sale of Products during such Calendar Quarter and shall distribute to RiconPharma its Profit Sharing Percentage of such Net Profits. ANI shall supply RiconPharma with a written report setting forth the Net Sales and Net Profits during such Calendar Quarter. If the Net Profits related to a Product for any Calendar Quarter is negative, then ANI may invoice RiconPharma for such amount due to ANI in accordance with this Agreement and in proportion to each Party’s Profit Sharing Percentage of such quarterly loss, such payment to be made within thirty (30) days of such invoice.
6. TAXES AND WITHHOLDING.
All payments under this Agreement shall be made without any deduction or withholding for or on account of any tax unless such deduction or withholding is required by applicable laws or regulations. If the paying Party is so required to deduct or withhold, such Party shall: (i) promptly notify the Party entitled to receive such payment of such requirement; (ii) pay to the relevant authorities the full amount required to be deducted or withheld promptly upon the earlier to occur of determining that such deduction or withholding is required or receiving notice that such amount has been assessed against the Party entitled to receive such payment; and (iii) promptly forward to such other Party an official receipt (or certified copy) or other documentation reasonably acceptable to such other Party evidencing such payment to such authorities. Unless otherwise required by law, each Party shall be responsible for the calculation and payment of its own taxes.
7. REGULATORY APPROVAL AND REGISTRATIONS.
7.1 ANI shall be responsible for obtaining all Regulatory Approvals for the Products necessary for the Registration of such Product in the Territory including the preparation of all ANDAs for the Products. ANI shall prepare all documents such as CMC and perform all other work necessary to obtain Regulatory Approvals and Registration of the Products in the Territory. The Parties will support the ANDA filing and help in obtaining Regulatory Approval for such Products, including, without limitation:
(a) seeking all necessary approvals to permit the conduct of Bioequivalence Studies using the Products;
(b) seeking all necessary Regulatory Approvals and Registrations from the appropriate Regulatory Authority in the Territory to manufacture, distribute, market and sell each of the Products in the Territory;
(c) preparing other applicable filings and obtaining approvals in connection with its advertising and promotional materials related to each of the Products; and
(d) the quality control testing of all Raw Materials used in the manufacture of each of the Products in accordance with the standards of the United States Pharmacopeia and any other specification which may be required by a Regulatory Authority;
(e) the pharmacokinetic and stability tests of the Products and the manufacture and scale-up of exhibit and registration stability Batches of the Products;
(f) the conduct of ongoing stability trials as required by any Regulatory Authority in the Territory; and
(g) the preparation and completion of any additional documentation necessary for the Registration of the Products in the Territory.
7.2 RiconPharma shall provide ANI with the identities of Raw Material sources including sources for the supply of approved APIs for the Products (“API Suppliers”). ANI will ensure that API Suppliers have maintained and, if required, filed appropriate DMFs in respect of the APIs used in the Products.
7.3 The testing and studies referred to in Sections 3.1 and 8.1 shall be conducted in accordance with all applicable GLPs and with all reasonable diligence. The Parties shall jointly review and comment upon, prior to submission, any documents submitted to any Regulatory Authority pertaining to the testing and study of a Product described in Sections 3.1 and 8.1 and DMFs covering any API in any Product.
7.4 No Party may attempt to modify the ANDA for a Product after Regulatory Approval in any way without the consent of the other Party.
7.5 Each Party shall immediately notify the other Party of any (a) inspections by any Regulatory Authority, including, without limitation, inspections as a result of the recall of, or any other regulatory issue related to, any of the Products and/or (b) material notices received from any Regulatory Authority. Each Party shall immediately notify the other Party if it becomes aware of any concern with respect to any Product that may affect the efficacy or safety of any of the Products.
8. MANUFACTURE AND SUPPLY OF THE PRODUCTS, QUALITY CONTROL.
8.1 ANI shall ensure that all Products supplied will be manufactured and packaged in accordance with GMPs and will conform to the Specifications, the applicable Regulatory Approval(s), and other regulations and requirements of the Regulatory Authorities.
8.2 ANI shall perform, or ensure the performance of, release testing of Products in a manner consistent with GMP testing methods agreed upon by the Parties as set forth on the Specifications. ANI shall provide RiconPharma with a Certificate of Analysis with each shipment of the Products stating that the Products in that shipment conform to the Specifications. ANI will ensure that a copy of the Certificate of Analysis with respect to each Batch of Product supplied (a) is faxed prior to shipping such Batch (confirmed by hard copies mailed) and (b) accompanies each Batch. ANI shall not ship any Batch of the Product if such Batch does not conform to the Specifications.
8.3 ANI shall ensure that each Batch of the Product is labeled and that each of the Batch numbers is applied to each such Batch, as required by the Regulatory Authorities.
8.4 ANI shall provide and maintain suitable storage and transport conditions for all Products shipped and shall provide complete written instructions with respect to proper conditions for the transport and storage of the Product. Upon receipt of any Batch of the Product from ANI, a Sales and Marketing Company shall provide and maintain storage conditions that comply with any written instructions provided by ANI in respect of the storage of Product.
9. PACKAGING AND LABELING.
ANI shall package and Label the Products shipped under this Agreement or ensure that such Products are packaged and Labeled in strict compliance with the Specifications and the packaging and Labeling requirements of the Regulatory Authorities. ANI shall be responsible for the accuracy and content of the Labeling.
10. REGULATORY REPORTING AND COMPLIANCE
10.1 Regardless of which Party or Parties hold the ANDA, ANI shall have the sole responsibility for monitoring and ensuring the material compliance with all statutes, regulations, guidelines and other requirements of the Regulatory Authorities pertaining to the Products, the Registration, and/or the
Regulatory Approval. ANI shall be responsible for ensuring appropriate work is performed with respect to supplementing or amending the approved ANDA and for complying with all reporting requirements relating to the Products and the Regulatory Approvals. Those duties include, without limitation, responding to physician questions regarding the Products and the submission of annual reports and adverse drug experience reports for the Products to the Regulatory Authority and the performance of all due diligence with respect to any adverse drug experience reports.
10.2 If RiconPharma receives a report or any other information regarding an adverse drug experience attributed to a Product, it shall promptly provide ANI with all such information. If either Party receives a communication from the Regulatory Authority regarding a Product, it will promptly notify the other Party in writing about that communication and shall provide a copy thereof. Upon request both ANI and RiconPharma shall provide each other with any other information they have or receive, if any, that ANI reasonably requires to comply with its obligations under Section 10.1.
10.3 Subject only to the specific duties imposed by Section 10.2, RiconPharma shall have no duties or responsibilities of any kind with respect to reporting to the Regulatory Authority or monitoring or ensuring compliance with any requirements of the Regulatory Authority.
11. RECALLS AND OTHER PRODUCT ACTIONS
11.1 Each Party shall promptly notify the other Party in writing of any order, request or directive of the Regulatory Authority or an order of a court to recall or withdraw a Product anywhere in the Territory (hereinafter “Recall”). The Party in whose name the ANDA is held shall be responsible for coordinating all communication in connection with the Recall, including all coordination and communications with the Regulatory Authority.
11.2 In the event that a Party believes it may be necessary to conduct a voluntary recall, field correction, market withdrawal, stock recovery or other similar action (hereinafter “Product Action”) with respect to any Product which was sold under this Agreement, such Party shall promptly consult with the other Party in good faith as to how best to proceed, it being understood and agreed that no Party shall be prohibited hereunder from taking any action that it is required to take by applicable law.
11.3 In the case of a Recall or Product Action, each Party shall make a permanent, complete and accurate record of all costs incurred by it in connection with the Recall or Product Action, a copy of which shall be delivered to the other Party as soon after the completion of such Recall or Product Action as may be practicable.
11.4 In the case of a Recall or Product Action that is covered by a recall insurance policy held by any Party, the Party holding the recall insurance policy shall be fully reimbursed the deductible amount in equal proportion by the Parties in accordance with the Profit Sharing Percentage for that Product. In the event that the recall expenses exceed insurance coverage, the Parties shall, except in the case of a Recall or Product Action that is covered by an indemnity obligation, share the expenses in accordance with the Profit Sharing Percentage for that Product.
11.5 In the case of a Recall or Product Action that is not covered by any indemnity obligation of a Party under Section 17 of this Agreement, the costs incurred by the Parties in connection with the Recall or Product Action shall be shared by the Parties in accordance with the Profit Sharing Percentage for that Product. In that event, if one Party has paid more than its share of the costs in connection with the Recall or Product Action, the other Parties, in accordance with their Profit Sharing Percentage for that Product, shall reimburse the overpaying Party the amount of the overpayment within sixty (60) days of receiving the record contemplated by Section 11.3.
12. ORDERS AND DELIVERY OBLIGATIONS.
12.1 The manufacturing Batch size of the Product that shall be listed on the corresponding Amending Product Exhibit. ANI or a Sales and Marketing Company designated by ANI shall accept orders for full-Batch quantities of the Product at the Base Price, as set forth in the corresponding Amending Product Exhibit.
12.2 The Amending Product Exhibit will set forth the Base Price for each Product. The Base Price may change from time to time based on the actual changes in the cost of Raw Materials, Components, labor, overhead, and costs related to stability testing and regulatory support services. ANI shall provide, at RiconPharma’s request, documentation illustrating how such Base Price changes have been calculated prior to the implementation of such change. Notwithstanding any other provision in this Agreement, ANI may not increase the labor and overhead elements of the Base Price calculation greater than [ *** ] without the express advance written consent of ANI and RiconPharma, which consent shall not be unreasonably withheld, conditioned or delayed.
13. RECORDS.
13.1 ANI will maintain records and documents documenting the Base Price of each Product and ANI will maintain records and documents documenting Net Sales and all transactions relating to the sale of each Product for a time period equal to the greater of:
(a) the period meeting all known regulations of the applicable Regulatory Authorities with respect to such Product; and
(b) five (5) years from the date of sale.
13.2 ANI shall maintain all records relating to the manufacture of the Products necessary to materially comply with all applicable laws, rules and regulations in each regulatory jurisdiction within the Territory and, if different, the regulatory jurisdiction of manufacture of the Products. Specifically, but without limitation, ANI shall maintain all records and samples (including retention samples) reasonably necessary to support GMPs and other regulatory requirements in such regulatory jurisdictions. All records relating to the manufacture of the Product shall be available for inspection, audit and copying by RiconPharma and its representatives and agents at ANI’s office upon reasonable advance request and during normal business hours. All such records shall be maintained for a period of not less than three (3) years or such longer period as may be required by law, rule or regulation. All records relating to the manufacture, stability and quality control of each Product shall be retained for a period of not less than the approved shelf life of such Product as set forth in the related Regulatory Approval plus two (2) years.
13.3 RiconPharma shall maintain all records relating to the development of the Products necessary to materially comply with all applicable laws, rules and regulations in each regulatory jurisdiction within the Territory. All records relating to the development of each Product shall be available for inspection, audit and copying by ANI and its representatives and agents at RiconPharma’s office upon reasonable advance request and during normal business hours. All such records shall be maintained for a period of not less than the period the Product is sold is by ANI or such longer period as may be required by law, rule or regulation.
14. INTELLECTUAL PROPERTY RIGHTS.
14.1 Each Party shall own all Inventions made solely by its employees and agents, and all patent applications and patents claiming such Inventions. All Inventions made jointly by employees or agents of the Parties and all patent applications and patents claiming such Inventions shall be owned jointly by the Parties. All determinations of invention under this Section 14 shall be in accordance with U.S. law
14.2 The Party owning the Invention shall make all decisions with respect to patent filings and shall have the right to select patent counsel and to take such other actions as are necessary to prepare, file, prosecute and maintain patent protection with regard to the Inventions under Section 14.1. With regard to jointly
owned Inventions, the Parties shall jointly determine in what countries, if any, patent applications claiming such joint Inventions should be filed. In the event that either Party does not wish to share in the expenses of filing, prosecuting or maintaining such joint Inventions in any country, such Party shall promptly assign or cause to be assigned to the other Party all of its right, title and interest in and to such joint Inventions in the subject country. Thereafter, such joint Invention shall be treated as an Invention solely owned by such other Party within the subject country for all purposes of this Agreement. In the event the Parties desire to proceed with the filing, prosecution and maintenance of such joint invention, they shall share in all expenses related thereto in accordance with each Party’s Development Cost Percentage on the Amending Product Exhibit for the Product to which the Invention relates or most closely relates.
14.3 Each Party shall be responsible for prosecuting and maintaining its own patent applications and patents. Except as otherwise provided in Section 14.2, all expenses for filing, prosecuting and maintaining a Party’s patent applications and patents shall be borne by such Party.
14.4 Each Party shall execute such documents as may be necessary to obtain, perfect or maintain any jointly owned patent rights filed pursuant to this Agreement. The Parties agree to cooperate with one another so far as reasonably necessary with respect to furnishing all information and data in its possession reasonably necessary to obtain or maintain such jointly owned patent rights.
14.5 Each Party shall have the sole right, in its own name and at its own expense, to enforce patent rights relating to Inventions that it owns against any Third Party. The Parties shall jointly determine which Party shall have the right and responsibility to institute, prosecute and control any action or proceeding with respect to the infringement or misappropriation of jointly owned patent rights.
14.6 In connection with any action taken by either Party against a Third Party to protect or enforce any patent right hereunder, the other Party shall, if requested, consult with the Party taking such action, and make its employees available as witnesses or as evidence any materials and/or data reasonably necessary for the furtherance of such action. Expenses incurred in connection with providing witnesses and/or making materials or data available shall be borne by the Party taking action against the Third Party.
14.7 If a Party is sued for infringing any Third Party patent out of the manufacture, use, sale or importation of a Product in the Territory, the Parties shall promptly discuss the course of action to be taken to resolve or defend such litigation. Each Party shall provide the other Parties with such assistance as is reasonably necessary and shall cooperate in the defense of such action.
15. RELATIONSHIP OF RICONPHARMA AND ANI.
15.1 The relationship of RiconPharma and ANI created by this Agreement is that of developer and contract manufacturer, and not that of a partnership, principal and agent, franchisor and franchisee, or joint or co-ventures. In the performance of this Agreement, no Party shall have any authority to assume or create any obligation or responsibility, either expressed or implied, on behalf of or in the name of any other Party, or to bind any other Party or its Affiliates in any manner whatsoever.
15.2 If this Agreement is terminated for any reason, the Parties shall not thereafter use, or permit anyone else under its control to use, any other Party’s name in the promotion of its business or the offer for sale of any goods.
16. REPRESENTATIONS AND WARRANTIES.
16.1 Each Party hereby represents and warrants to the other Parties that:
(a) it is a corporation or other entity duly organized and validly existing under the laws of the state or other jurisdiction of its incorporation or organization;
(b) the execution, delivery and performance of this Agreement by such Party has been duly authorized by all requisite corporate or other entity action and does not require any shareholder or member action or approval;
(c) it has the power and authority to execute and deliver this Agreement and to perform its obligations hereunder;
(d) the execution, delivery and performance by such Party of this Agreement and its compliance with the terms and provisions hereof does not and will not conflict with or result in a breach of any of the terms and provisions of or constitute a default under: (i) a loan agreement, guaranty, financing agreement, agreement affecting a Product or other agreement or instrument affecting a Product; (ii) the provisions of its charter or other operative documents or bylaws; or (iii) any order, writ, injunction or decree of any court or governmental authority entered against it or by which any of its property is bound;
(e) it has the full right, power and authority to grant all of the right, title and interest in the licenses, if any, granted to the other Party under this Agreement;
(f) it is financially solvent and has the financial resources to perform its obligations under this Agreement; and
(g) it is not debarred under the Generic Drug Enforcement Act of 1992; it does not and will not use in any capacity the services of any person debarred under the Generic Drug Enforcement Act of 1992; and neither it nor any of its employees, agents, suppliers, or contractors has engaged in any activity which could lead to it becoming debarred under the Generic Drug Enforcement Act of 1992.
16.2 ANI covenants, represents, and warrants that:
(a) it shall at all times comply with all material applicable laws, rules, and regulations relating to its activities under this Agreement;
(b) all Products shipped shall be manufactured, packaged, stored, and shipped by ANI materially in accordance with all GMPs and all other applicable laws, rules and government regulations in effect at the time of shipping of the Product;
(c) the Labeling content for the Products will at all times comply with the Regulatory Approval for the Product and be the same as the Labeling for the Reference Product except for differences allowed by applicable regulations;
(d) all Products shipped shall conform to the Specifications, the Regulatory Approval, and the Registration and be merchantable and shall not be misbranded or adulterated;
(e) it shall take all commercially reasonable precautions customary in the industry in manufacturing, testing, packaging, labeling and handling the Products to ensure the quality, safety and fitness thereof;
(f) that each Unit of the Products shall bear an expiration date of no less than twenty-four (24) months following the date of its manufacture, unless the RLD has an expiration date of less than twenty-four (24) months, in which case the expiration date borne by the Products shall reflect the stability of the RLD;
(g) it shall materially comply with all requirements of the laws and regulations of the Regulatory Authority and applicable state law requirements governing the marketing, sale and distribution of Products;
(h) it shall maintain adequate warehousing, distribution facilities, documentation and personnel to provide reasonable distribution, staffing for customer service, billing, marketing and accounting with respect to the Products; and
(i) copies of all agreements ANI has with any Third Party or Affiliate for manufacture or supply of any Product, Raw Materials or Components in effect on the date hereof are attached as exhibits to this Agreement and the copies attached as exhibits are true and complete copies of those agreements.
16.3 RiconPharma covenants, represents, and warrants that:
(a) it shall at all times comply with all applicable laws, rules, and regulations relating to its activities under this Agreement;
(b) based on its preliminary analysis, RiconPharma believes that Commercially Reasonable Efforts will result in the ability to develop and manufacture and obtain Regulatory Approval for the Products;
(c) all of the research and development activities pertaining to the Products shall be conducted in accordance with all applicable laws and regulations, GMPs, GLPs and GCP and all applicable guidelines promulgated by any Regulatory Authority having jurisdiction over the Products in the Territory;
(d) copies of all agreements RiconPharma has with any Third Party or Affiliate for manufacture or supply of any Product, Raw Materials or Components in effect on the date hereof are attached as exhibits to this Agreement and the copies attached as exhibits are true and complete copies of those agreements.
17. INDEMNIFICATION
17.1 RiconPharma shall indemnify, defend, and hold harmless ANI and its respective Affiliates, directors, officers, owners, employees, and agents from and against any and all claims, demands, lawsuits, causes of action, actions or other proceedings, losses, liabilities, injuries, damages, and expenses, including reasonable attorneys’ fees and costs from any actual or alleged infringement of patent, trademark, trade dress or other intellectual property rights or interests of any Third Party by the Products, the sale of the Products and/or the development of the Products. The indemnity obligations in the preceding sentence shall not apply to an actual infringement caused solely by the willful misconduct of ANI.
17.2 ANI shall indemnify, defend, and hold harmless RiconPharma and their respective Affiliates, directors, officers, owners, employees, and agents from and against any and all claims, demands, lawsuits, causes of action, actions or other proceedings, losses, liabilities, injuries, damages, and expenses, including reasonable attorneys’ fees and costs from any actual or alleged infringement of patent, trademark, trade dress or other intellectual property rights or interests of any Third Party by the Labeling of the Products and/or the methods, design, or processes utilized in connection with the manufacturing or packaging of the Products. The indemnity obligations in the preceding sentence shall not apply to an actual infringement caused solely by the willful misconduct of RiconPharma.
17.3 RiconPharma shall indemnify, defend, and hold harmless ANI and their respective Affiliates, directors, officers, employees, owners and agents from and against any and all claims, demands, lawsuits, causes of actions, actions or other proceedings (including voluntary and involuntary Product Recalls, Product
Actions, and other FDA enforcement actions), losses, liabilities, injuries, damages and expenses, including reasonable attorneys’ fees and costs arising from or relating to: (i) the breach of any representation, warranty or covenant made or given by RiconPharma in this Agreement; (ii) any breach of any obligations under Section 10 of this Agreement; (iii) the design or formulation of any Products including, but not limited to, any defect in design, whether patent or latent; and/or (iv) any negligent act or omission by RiconPharma or its Affiliates.
17.4 ANI shall indemnify, defend, and hold harmless RiconPharma and its respective Affiliates, directors, officers, employees, owners and agents from and against any and all claims, demands, lawsuits, causes of actions, actions or other proceedings (including voluntary and involuntary Product Recalls, Product Actions, and other FDA enforcement actions), losses, liabilities, injuries, damages and expenses, including reasonable attorneys’ fees and costs arising from or relating to: (i) the breach of any representation, warranty or covenant made or given by ANI in this Agreement; (ii) any defect in Raw Materials or Components that ANI knew or reasonably should have known through testing in compliance with GMPs, including, but not limited to, any defect from or relating to the handling, storage, formulation, testing, supply, packaging, purchase, or manufacture of any Raw Materials or Components; (iii) the handling or storage by ANI of any Products; (iv) the manufacture, testing, or packaging of any Products including, but not limited to, any defect in manufacturing; (v) the Labeling of the Products including, but not limited to, any defect in warning; and (vi) any grossly negligent act or omission by ANI or its Affiliates.
17.5 A Party shall give written notice to the other Party of a claim, demand, lawsuit, cause of action, action or other proceeding (including voluntary and involuntary Product Recalls, Product Actions, and other FDA enforcement actions), loss, liability, injury, damage and/or expense, including reasonable attorneys’ fees and costs (hereinafter individually and collectively a “Claim”) for which the Party contends it is entitled to be defended, indemnified and held harmless under Sections 17.1 through 17.4 (hereinafter individually and collectively an “Indemnity Claim”) within 30 days after the Party making the Indemnity Claim becomes aware of the Claim; provided, however, that the failure to give such notice within the 30 day period shall not waive or in any way impair a Party’s right to be indemnified, defended or held harmless unless the delay in providing such notice has a material adverse effect on the indemnifying Party. An Indemnity Claim shall be deemed accepted unless within 30 days after receiving the Indemnity Claim notice, the receiving Party notifies the other Party in writing that the receiving Party will not defend, indemnify and hold the sending Party harmless
17.6 An indemnified Party shall reasonably cooperate with the indemnifying Party with respect to any investigation or defense of any Claim.
17.7 Upon receiving notice of a Claim, ANI may, for purposes of funding reserves for Litigation Expenses and related damages, withhold Net Profits that would otherwise be distributed to RiconPharma pursuant to Section 5.2, place such amounts in escrow and use such reserves to pay Litigation Expenses from a Claim or to pay the deductible or SIR and related damages on any Policy.
17.8 The indemnifying Party shall have the right to control the defense or settlement of an Indemnity Claim that it has accepted. The indemnified Party may participate in (but not control) the defense of a Claim at its sole cost and expense.
17.9 Where a Party refuses to accept an Indemnity Claim, the other Party’s defense of the Claim will not be deemed a waiver or admission of any kind against the refusing Party for indemnity, defense and to be held harmless under this Section 17.
17.10 A Party may not settle a Claim without the consent of another Party [if such settlement would require the other Party to submit to an injunction].
18. INSURANCE.
18.1 Not later than the Initial Marketing Date with respect to a Product, each Party shall obtain and maintain during the Term of this Agreement and for a period of three years after the Termination of this Agreement at its own expense insurance policies, including product liability insurance, providing coverage for any personal injury or property damage allegedly caused by the Products or the acts and omissions of the Parties relating to the Products (the “Policies”) with liability limitations of: (i) in the cases of ANI, not less than [ *** ] per occurrence and in the aggregate; and (ii) in RiconPharma’s case [ *** ] per occurrence and in the aggregate if there is one Amending Product Exhibit to this Agreement. If there is more than one Amending Product Exhibit to this Agreement, or if the total dollar value of sales of Products by ANI exceeds [ *** ], the liability limitations for the RiconPharma Policies shall be a minimum of [ *** ] per occurrence and in the aggregate. Each Party shall deliver a certificate of insurance on an Accord form or equivalent to the other Party evidencing that the Policies are in effect and providing that the Policies will not be cancelled or modified without first giving 30 days advance notice to the certificate holder. Any Party may request to be named as an additional insured on the Policies obtained and maintained by the other Party, subject to approval by the insurance provider. The Policies obtained and maintained by each Party shall provide contractual liability insurance providing coverage for the indemnity, defense and hold harmless obligations undertaken by that Party under this Agreement.
18.2 Subject only to Section 18.3 below, the Policies obtained and maintained by RiconPharma shall be primary to any Policies obtained and maintained by ANI with respect to any Claim. The Policies shall have a maximum deductible or self-insurance retention (“SIR”) of [ *** ] per policy.
18.3 ANI’s Policies shall be primary with respect to RiconPharma’s Policies as to any Claim for which ANI owes indemnity and defense to RiconPharma under Section 17.4 of this Agreement.
19. AUDITS AND INSPECTIONS.
19.1 Each Party shall keep complete and accurate accounts, records, books and-data with respect to its obligations hereunder including the books and records identified and described in Section 13 of this Agreement (the “Records”). Each Party shall have the right at reasonable times upon prior written notice to another Party, to inspect copy and audit the Records relating to the other Party’s performance and obligations under this Agreement. A Party shall permit authorized representatives of the other Party to inspect the Party’s facilities and quality systems in connection with the Product during standard business hours and for reasonable periods for the purpose of assuring that the Party is complying with the federal and state laws and regulations relating to the production of the Product. Such inspection shall be at the inspecting Party’s sole expense and upon at least five (5) business days advance written notice to the other Party.
19.2 In the event of an audit or inspection of one of the Party’s facilities by any Regulatory Authority, that Party shall supply the other Parties with notice of the audit or inspection and a copy of any report received from such Regulatory Authority and the inspected Party shall provide such Regulatory Authority with a prompt, accurate and complete response to any deficiencies or observations noted during the audit or inspection. The Party inspected or audited agrees that it shall promptly address, and if necessary correct, any and all such deficiencies or observations, and obtain any required approval or reclassification from the Regulatory Authority.
19.3 In the event of an audit or inspection of one of the facilities of a Third Party manufacturer or supplier of any Raw Materials or Components, the Party who hired or contracted with the Third Party (the “Retaining Party”) shall notify the other Party of the audit or inspection and provide to the other Parties a copy of any report the Retaining Party receives issued by the Regulatory Authority pertaining to the audit or inspection.
20. TERM AND TERMINATION.
20.1 This Agreement shall become effective on the Effective Date and continue until terminated in accordance with this Agreement (the “Term”).
20.2 The Parties may jointly agree in writing, at any time, to terminate any Product Exhibit to this Agreement. In the event that any Amending Product Exhibit to this Agreement is terminated pursuant to this Section 20.2, the effect of such termination shall be as set forth in the agreement between the Parties documenting that termination. In the event that all Amending Product Exhibit to this Agreement have been terminated, a Party may terminate this Agreement by providing the other Parties with thirty (30) days’ prior written notice.
20.3 This Agreement or any Amending Product Exhibit may be terminated effective immediately by written notice by a Party to the other Party at any time during the Term of this Agreement for material breach by the other Party of any provision of this Agreement, which breach remains uncured for thirty (30) days from the date written notice of such breach is given to the breaching Party; provided, however, that if such breach is not cured within the stated period and the breaching Party uses Commercially Reasonable Efforts to cure such breach, the stated period will be extended by an additional thirty (30) days.
20.4 Prior to the issuance of an ANDA for a Product, a Party may terminate its interest and involvement in the Amending Product Exhibit for that Product upon thirty (30) days’ prior written notice to the other Party. In that event, the terminating Party shall be required to pay or reimburse the non-terminating Party for its respective share of the Development Costs as set forth in the Amending Product Exhibit. In addition, the terminating Party shall not pursue the development, manufacture, marketing, distribution or sale of such Product or a Bioequivalent Product manufactured by a Third Party for a period of five (5) years after the effective date of such termination.
20.5 After the issuance of an ANDA for a Product, a Party that desires to terminate its interest or involvement in an Amending Product Exhibit to this Agreement with respect to such Product other than by reason of Section 20.3, shall give the other Parties not less than sixty (60) days’ prior written notice thereof, such termination to be effective at the conclusion of such sixty (60) day period.
20.6 If a Party terminates its interest in an Amending Product Exhibit to this Agreement by reason of Sections 20.4 or 20.5, or the other Party terminates this Agreement or any Amending Product Exhibit pursuant to Section 20.3, the Party not terminating or in breach, as applicable (the “Continuing Party”), may continue to develop, market, manufacture, and sell the Product on the Amending Product Exhibit, as the case may be, and shall have the right to purchase the rights of the breaching or terminating Party as the case may be (the “Non-Continuing Party”) in and to the Product. The Non-Continuing Party shall contemporaneously assign or license, as applicable, to the Continuing Parties, all of the Non-Continuing Party’s rights (including proprietary rights) to continue to develop, make, have made, use, import, market, offer for sale or sell the Products, including any Manufacturer and Development Technology. The assigned or licensed rights, as applicable, shall include, without limitation, Regulatory Approvals (including the ANDA), the trade name for the Product and the manufacturing rights, the good will related to the Product, accounts receivable, and inventory of the Product. The purchase price of such assigned or licensed rights shall be [ *** ] and the following additional provisions shall apply:
(a) By ANI. If ANI is the Non-Continuing Party then, subject to the terms and conditions of this Agreement, (i) it shall grant to RiconPharma non-exclusive license in and to Manufacturing and Development Technology it has to the extent necessary for RiconPharma to perform ANI’s obligations and exercise its rights under this Agreement, including, without limitation, any and all obligations and rights that need to be performed or exercised by or on behalf of ANI to develop, make, have made, use, import, market, offer for sale or sell the Product in accordance with this Agreement and (ii) at the request of RiconPharma, ANI shall manufacture the Product in compliance with the terms and conditions of this Agreement for a period not to exceed twelve (12) months following the notice of termination. Following any termination in respect of which ANI is the Non-Continuing Party, ANI shall use Commercially Reasonable Efforts to assist RiconPharma in transferring the manufacturing of the Product to a Third Party. For a period of twelve (12)
months following the notice of termination, ANI shall not develop, apply for Regulatory Approval for, manufacture, import, market, sell or promote any product that is a direct substitute for the Product, including any Bioequivalent Product. If the ANDA is in ANI’s name, it shall execute all documents and take all other actions necessary to transfer the ANDA in accordance with the instructions of ANI and RiconPharma,
(b) By RiconPharma. If RiconPharma is the Non-Continuing Party then, subject to the terms and conditions of this Agreement, it shall grant to ANI non-exclusive license in and to Manufacturing and Development Technology it has to the extent necessary for ANI to perform RiconPharma’s obligations and exercise its rights under this Agreement, including, without limitation, any and all obligations and rights that need to be performed or exercised by or on behalf of RiconPharma to develop, make, have made, use, import, market, offer for sale or sell the Product in accordance with this Agreement. For a period of twelve (12) months following the notice of termination, RiconPharma shall not develop, apply for Regulatory Approval for, manufacture, import, market, sell or promote any product that is a direct substitute for the Product, including any Bioequivalent Product. If the ANDA is in RiconPharma’s name, it shall execute all documents and take all other actions necessary to transfer the ANDA in accordance with the instructions of ANI.
21. CONFIDENTIALITY.
During the term of this Agreement, each Party will be exposed to confidential proprietary technical information belonging to the other Party that pertains to the operation of the other Party’s businesses or the operation of its business in general, including but not limited to the formulations, related technical information and data, packaging, research, operations, manufacturing processes, marketing, strategy, know-how and product information for a Product. Each Party agrees not to (i) disclose, during the term of this Agreement or thereafter, to any other person any Confidential Information of the Party, or (ii) use, after the term of this Agreement, any Confidential Information of the other Party for any purpose. “Confidential Information” includes all information that derives independent economic value from not being generally known, or not being readily ascertainable by proper means, by other persons who can obtain economic value from the disclosure or use of such information, and any other information identified by a Party as being Confidential Information. For purposes of this Agreement, “Confidential Information” does not include any information that (i) at the time of disclosure or thereafter is publicly available (other than as a result of a violation of this Paragraph), (ii) was or becomes available to the recipient on a non-confidential basis from a source other than the disclosing Party, provided that such source is not and was not bound by a confidentiality agreement with or other obligation of secrecy to the disclosing Party; or (iii) is independently acquired or developed by the recipient without violating any of its obligations under this Paragraph. The obligations of the Parties set forth in this Paragraph shall survive the termination or expiration of this Agreement.
22. FORCE MAJEURE.
No Party shall be liable or be in breach of any provision of this Agreement for any failure or delay on its part to perform any obligation where such failure or delay has been occasioned by any act of God, war, riot, fire, explosion, flood, sabotage, unavailability of fuel, labor, containers or transportation facilities, accidents of navigation or breakdown or damage of vessels or other conveyances for air land or sea, other impediments or hindrances to transportation, government intervention (other than that of a duly-authorized Regulatory Authority), strikes or other labor disturbances or any other cause beyond the control of the Parties.
23. NOTICES.
All notices and other communications required or permitted to be given or made pursuant to this Agreement shall be in writing signed by the sender and sent to the address or number below by facsimile transmission or Federal Express or another recognized overnight mail service that utilizes a written form of receipt for next day or next business day delivery. The notice shall be deemed duly given (a) if faxed by
4:00 p.m., New York time, on the date sent by fax provided there is a confirmation by the transmitting machine showing the proper number of pages were transmitted without error or (b) if sent by overnight mail, on the business day after being sent by Federal Express or another recognized overnight mail service which utilizes a written form of receipt for next day or next business day delivery. A Party may change its address or fax number for receiving notice by the proper giving of notice hereunder:
To: ANI
ANI Pharmaceuticals, Inc.
210 Main Street West
Baudette, MN 56623
USA
Attention: Vice President & CFO
Fax: +1(218) 634-3540
To: RiconPharma
RiconPharma LLC
100 Ford Road, Suite #9
Denville, NJ 07834
USA
Attention: President & CEO
Fax: +1(973) 627-4735
24. EXECUTION OF ALL NECESSARY ADDITIONAL DOCUMENTS.
Each Party agrees that it will forthwith upon the request of the other Party execute and deliver all documents and will take all such other actions as the other Party may reasonably request from time to time in order to effectuate the provision and purposes of this Agreement.
25. WAIVER.
Any failure of a Party to enforce at any time any of the provisions of this Agreement shall not be deemed or construed to be a waiver of such provisions or a waiver of any right of such Party thereafter to enforce each and every such provision on any succeeding occasion or breach thereof.
26. ASSIGNMENT AND AMENDMENT.
26.1 Other than an assignment pursuant to Section 2.3, neither this Agreement nor any rights hereunder shall be assigned by a Party without the prior written consent of the other Party, and then only upon approval of the other Party and acceptance of such assignment in written form approved by such Party, which approval shall not be unreasonably withheld, conditioned or delayed. In the event of an assignment by a Party to any Affiliate thereof as permitted hereunder, the assigning Party shall not be released from its obligations hereunder, and shall guarantee the full performance by such Affiliate of such obligations.
26.2 No amendment hereof shall be binding unless made in writing and signed by each of the Parties hereto.
27. ENTIRE AGREEMENT.
This Agreement, including the Amending Product Exhibits to this Agreement, incorporates the entire understanding of the Parties and revokes and supersedes any and all agreements, contracts, understandings or arrangements that might have existed heretofore among the Parties regarding the subject matter hereof, and all prior agreements and understandings between the Parties and relating to the subject matter hereof are superseded by this Agreement. No Party shall be liable or bound to another Party in any manner by any representations, warranties or covenants relating to such subject matter except as specifically set forth herein. This Agreement may be executed in any number of counterparts, each of which shall be an original,
but all of which together shall constitute one instrument. This Agreement may also be executed via facsimile, which facsimile shall be deemed an original.
28. GOVERNING LAW; ARBITRATION; LANGUAGE.
28.1 This Agreement shall be governed by and interpreted in accordance with the substantive laws of the State of New York, without giving effect to conflict of law principles thereof, and the Parties consent to and agree to submit to the jurisdiction of the courts of, and accept service of process from, the State of New York, state and federal, with respect to any Claim or other claim, action, lawsuit, or proceeding relating to or out of this Agreement. The Parties expressly agree that, to the maximum extent permitted by law, the requirements of any multilateral or bilateral treaties, now or hereafter existing, between two or more countries that place any obligations or duties on a Party or the Parties that are inconsistent with or in addition to any of its obligations and duties under this Agreement, shall not apply to this Agreement or to the Parties’ performance hereunder without the consent of all Parties. This Agreement shall exclude, and not be governed by, either the provisions of the International Sale of Goods Act, or the United Nations Convention on the International Sale of Goods, regardless of that Convention’s legal or statutory adoption by any jurisdiction.
28.2 In the event of any Indemnity Claim or other dispute, claim, question or disagreement out of or relating to this Agreement, or the breach hereof, the Parties shall use Commercially Reasonable Efforts to settle such Indemnity Claim or other dispute, claim, question or disagreement. To this end, the Parties shall consult and negotiate with each other in good faith and, recognizing mutual interests, attempt to reach a just and equitable solution satisfactory to each of the Parties. If the Parties do not reach such resolution within a period of thirty (30) days, any Indemnity Claim or other dispute, claim, question or disagreement out of or relating to this Agreement, or the breach hereof, where the total amount in controversy between the Parties is less than [ *** ], shall be determined and settled by binding arbitration in New York County, New York before a three member panel of the American Arbitration Association or JAMS in accordance with the provisions of the tribunal’s then applicable Commercial Arbitration Rules. Notice of the demand for arbitration shall be made in writing to the other Party and to the arbitral tribunal. Nothing contained in this Section 28.2 shall prevent a Party from seeking interim or final equitable relief from the arbitral tribunal of a state or federal court of competent jurisdiction in the State of New York. In no event shall the demand for arbitration be made after the date when institution of legal or equitable proceedings based on the claim or dispute would be barred by the applicable statute of limitations. Any award rendered by the arbitration panel shall be final and conclusive upon the Parties and a judgment thereon may be entered in any court having competent jurisdiction.
28.3 Each Party represents that it has been represented by legal counsel in connection with this Agreement. In interpreting and applying the terms and provisions of this Agreement, the Parties agree that no presumption shall exist or be implied against the Party which drafted such terms and provisions.
29. SEVERABILITY.
If any term or provision of this Agreement shall be held invalid or unenforceable, the remaining terms hereof shall not be affected, but shall be valid and enforced to the fullest extent permitted by law.
30. HEADINGS.
The headings used in this Agreement are intended for guidance only and shall not be considered part of this written understanding between the Parties hereto and shall have no effect on the meaning of the provisions hereof.
31. SURVIVAL.
All representations, warranties, and covenants of the Parties and the terms and conditions of Sections 3.2, 5, 10, 11, 14, 17, 18, 21, and 28 shall survive the termination of any Amending Product Exhibit and/or this Agreement, notwithstanding any language in the Agreement to the contrary.
[remainder of page intentionally left blank; signature page follows]
IN WITNESS WHEREOF, this Agreement has been executed by the Parties on the date first above written.
ANIP Acquisition Company | |||
By: | /s/ Charlotte C. Arnold | ||
Name: | Charlotte C. Arnold | ||
Title: | Vice President & CFO | ||
RiconPharma LLC | |||
By: | /s/ Raj Devalapalli | ||
Name: | Raj Devalapalli | ||
Title: | President & CEO | ||
AMENDING PRODUCT EXHIBIT A-1
[ *** ]
AMENDING PRODUCT EXHIBIT A-2
[ *** ]
AMENDING PRODUCT EXHIBIT A-3
[ *** ]
AMENDING PRODUCT EXHIBIT A-4
[ *** ]
AMENDING PRODUCT EXHIBIT A-5
[ *** ]
Exhibit 10.26
Execution Draft
EXECUTIVE EMPLOYMENT AGREEMENT
EXECUTIVE EMPLOYMENT AGREEMENT (this “Agreement”) between ANI Pharmaceuticals, Inc. (the “Company”) and Christopher Mutz (“Executive”) dated as of February 10, 2021. Each of the Company and Executive are sometimes referred to herein individually as a “Party” and together as the “Parties.”
WHEREAS, the Company wishes to employ Executive, and Executive wishes to be employed by the Company on the terms and conditions set forth herein.
NOW, THEREFORE, in consideration of the mutual promises, terms, covenants and conditions set forth herein and the performance of each, the Parties hereto, intending to be legally bound, hereby agree as follows:

4148-1903-4155.4
2
3
4
5
6
(A) continue to pay Executive his then current Base Salary, payable in regular installments in accordance with the Company’s standard payroll procedures but no less frequently than bi-monthly procedures for a period equal to twelve (12) months following the Termination Date (the “Severance Period”);
7
(B) if Executive elects to receive continuation coverage under the Consolidated Omnibus Budget Reconciliation Act of 1985, as amended (“COBRA”), pay Executive on a monthly basis for the Severance Period, an amount equal to a percentage (based on the portion of the monthly premium costs covered by the Company for Executive’s group health, dental and/or vision coverage in effect as of the Termination Date), of Executive’s monthly COBRA premium payment (if any) under the Company’s group health, dental and vision plans; provided, however, that the obligations of the Company under this clause (B) shall cease upon Executive becoming eligible to participate in a plan of another employer providing substantially similar group health benefits to Executive and his eligible family members and dependents or upon termination of Executive’s COBRA coverage;
(C) if such termination occurs after June 30th in any calendar year, pay Executive a pro-rated Incentive Bonus for the fiscal year during which Executive’s employment is terminated (prorated based on the days elapsed in such fiscal year through the Termination Date);
(D) a lump sum cash payment equal to the Executive’s annual maximum Incentive Bonus Amount, payable on the first payroll date following the first anniversary of the Termination Date (all of the payments in clauses (A) - (D) together, the “Severance Payments”); and
(E) all of the Executive’s options to purchase the common stock of the Company and any awards of restricted common stock received by Executive in each case that were subject to vesting shall vest with respect to that number of shares subject thereto that would have vested during the Severance Period had Executive remained employed by the Company during such period, and such vested options (after taking into account the vesting acceleration) shall remain exercisable through the eighteen (18) month anniversary of the Termination Date (but in no event beyond the original term of the equity award), it being understood that to the extent any options that are intended to be “incentive stock options” for purposes of Section 422 of the Internal Revenue Code are exercised after the three month anniversary of the Termination Date, such options will cease to be treated as “incentive stock options” for purposes of Section 422 of the Internal Revenue Code.
8
9
10
(A) With respect to the Generic Company Products, the development, manufacturing, marketing, distribution or sale, or licensing of any pharmaceutical product in the Territory (1) that is an AB-rated
11
generic equivalent of the same Reference Listed Drug product as a Generic Company Product and for which there are four or fewer AB-rated generic equivalent competitors or (2) that is a direct competitor of any Company Product that is a DESI Product and that utilizes the same active pharmaceutical ingredients as such DESI Product;
(B) With respect to the Brand Company Products, the development, manufacturing, marketing, distribution or sale, or licensing of: (1) any pharmaceutical product in the Territory that is an AB-rated generic equivalent of a Brand Company Product and (2) for which either (I) there are three or fewer AB-rated generic equivalent competitors or (II) AB-rated generic equivalents have been available for commercial sale in the Territory for no more than three years; and
(C)The development, manufacturing, marketing, distribution or sale, or licensing of any product utilizing Corticotropin as the active pharmaceutical ingredient.
12
13
For purposes hereof, “Confidential Information” means and means any and all confidential, proprietary or Trade Secret information of the Company or its controlled affiliates not within the public domain, whether disclosed, directly or indirectly, verbally, in writing (including electronically) or by any other means in tangible or intangible form, including that which is conceived or developed by Executive, applicable to or in any way related to: (i) the present or future business activities, products and services, and customers of the Company or its controlled affiliates; (ii) the research and development of the Company or its controlled affiliates; or (iii) the business of any customers or suppliers of the Company or its controlled affiliates. Such Confidential Information includes the following property or information of the Company or its controlled affiliates, by way of example and without limitation, trade secrets, processes, formulas, data, program documentation, customer lists, pricing information, designs, drawings, algorithms, source code, object code, technology, formulae, models, know-how, improvements, pharmaceutical drug and/or devise technologies, inventions, licenses, techniques, all plans or strategies for marketing, development and pricing, government filings and/or reports, inventions, research, development, schematics, designs, test methods and samples, documents, agreements, business plans, financial statements, profit margins and all information concerning existing or potential clients, suppliers or vendors. Confidential Information of the Company also means all similar information disclosed to the Company by third parties that is subject to confidentiality obligations.
The Company shall not be required to advise Executive specifically of the confidential nature of any such information, nor shall the Company be required to affix a designation of confidentiality to any tangible item, in order to establish and maintain its confidential nature. Notwithstanding the preceding to the contrary, Confidential Information shall not include general industry information or information that is publicly available or readily discernable from publicly available products or literature; information that the Executive lawfully acquires from a source other than the Company or its controlled Affiliates or any customer or supplier of the Company or any of its controlled Affiliates (provided that such source is not bound by a confidentiality agreement with the Company or any of its controlled Affiliates); information that is required to be disclosed pursuant to any law, regulation, rule of any governmental body or authority, or stock exchange, or court order; or information that reflects Executive’s own skills, knowledge, know-how and experience gained prior to employment or service and outside of any connection to or relationship with the Company or any of its controlled Affiliates, or the predecessors of any such entities.
For purposes hereof, the term “Trade Secret” shall have the meaning given in the Delaware enactment of the Uniform Trade Secrets Act, and shall include, without limitation, the whole or any portion or phase of any scientific or technical information, design, process, formula, concept, data organization, manual, other system documentation, or any improvement of any thereof, in any case that is valuable and secret (in the sense that it is not generally known to the Company’s competitors).
Notwithstanding the foregoing, the U.S. Defend Trade Secrets Act of 2016 (“DTSA”) provides that an individual shall not be held criminally or civilly liable under any federal or state trade secret law for the disclosure of a trade secret that is made (i) in confidence to a federal, state, or local government official, either directly or indirectly, or to an attorney; and (ii) solely for the
14
purpose of reporting or investigating a suspected violation of law; or (iii) in a complaint or other document filed in a lawsuit or other proceeding, if such filing is made under seal. In addition, DTSA provides that an individual who files a lawsuit for retaliation by an employer for reporting a suspected violation of law may disclose the trade secret to the attorney of the individual and use the trade secret information in the court proceeding, if the individual (A) files any document containing the trade secret under seal; and (B) does not disclose the trade secret, except pursuant to court order.
15
16
17
(A) one Person (or more than one Person acting as a group) acquires (or has acquired during the 12-month period ending on the date of the most recent acquisition by such Person or group) assets of the Company (including its subsidiaries) that have a total gross fair market value equal to more than fifty percent (50%) of the total gross fair market value of all of the assets of the Company immediately before such acquisition or acquisitions, or
(B) one Person, or more than one Person acting as a group, acquires ownership of equity securities of the Company (including by way of merger, consolidation or otherwise) that, together with all equity securities of the Company previously held by such Person or group, constitutes more than fifty percent (50%) of the total fair market value or total voting power of equity securities of the Company.
Notwithstanding the foregoing, a Change in Control shall not include (1) any transaction effected for reincorporation purposes or (2) any transaction that does not constitute a change of ownership of the Company or a substantial portion of its assets within the meaning of Treasury Regulation Section 1.409A-3(i)(5)(v) or (vii).
(A)(1) a Termination of Employment by the Company has occurred for any reason other than for Good Cause or (2) a Termination of Employment by Executive for Good Reason has occurred; and
(B)A Change in Control has been consummated; and
(C)the Termination of Employment, has occurred either: (1) within the period beginning on the date of the consummation of the Change in Control and ending on the last day of the twenty-fourth (24th) month following the consummation of a Change in Control; or (2) prior to a Change in Control, if such Termination of Employment was either a condition of the Change in Control or was at the documented request or insistence of a Person which is a party or an Affiliate of a party to the transaction that constitutes or results in to the Change in Control.
18
19
20
To the Company:
ANI Pharmaceuticals, Inc.
210 Main Street West
Baudette, MN 56623
Attn: General Counsel or the Chief Financial Officer
Telephone No.: 218-634-3500
E-Mail: stephen.carey@anipharmaceuticals.com
To Executive:
Christopher Mutz
at the contact information on file with the Company
Either party may, by notice given in accordance with this Section, specify a new address for
21
notices under this Agreement.
22
[Signature Page Follows]
23
IN WITNESS WHEREOF, the Parties hereto have executed this Executive Employment Agreement as of the day and year first above written.
Company:
ANI PHARMACEUTICALS, INC.
Name: Nikhil Lalwani
Title: Chief Executive Officer
Executive:
/s/ Christopher Mutz
Christopher Mutz
Exhibit A: General Release of All Claims
Exhibit B: Indemnification Agreement
[SIGNATURE PAGE TO EXECUTIVE EMPLOYMENT AGREEMENT]
4148-1903-4155.4
EXHIBIT A
GENERAL RELEASE OF ALL CLAIMS
[See Attached]
4148-1903-4155.4
EXHIBIT B
INDEMNIFICATION AGREEMENT
[See Attached]
4148-1903-4155.4
Exhibit 10.27
Execution Version
EXECUTIVE EMPLOYMENT AGREEMENT
EXECUTIVE EMPLOYMENT AGREEMENT (this “Agreement”) between ANI Pharmaceuticals, Inc. (the “Company”) and Ori Gutwerg (“Executive”) dated as of January 15, 2021. Each of the Company and Executive are sometimes referred to herein individually as a “Party” and together as the “Parties.”
WHEREAS, the Company wishes to employ Executive, and Executive wishes to be employed by the Company on the terms and conditions set forth herein.
NOW, THEREFORE, in consideration of the mutual promises, terms, covenants and conditions set forth herein and the performance of each, the Parties hereto, intending to be legally bound, hereby agree as follows:
4124-3184-4650.4
2
3
4
5
6
(A) continue to pay Executive his then current Base Salary, payable in regular installments in accordance with the Company’s standard payroll procedures but no less frequently than bi-monthly procedures for a period equal to twelve (12) months following the Termination Date (the “Severance Period”);
(B) if Executive elects to receive continuation coverage under the Consolidated Omnibus Budget Reconciliation Act of 1985, as amended (“COBRA”), pay Executive on a monthly basis for the Severance Period, an amount equal to a percentage (based on the portion of the monthly premium costs covered by the Company for Executive’s group health, dental and/or vision coverage in effect as of the Termination Date), of Executive’s monthly COBRA premium payment (if any) under the Company’s group health, dental and vision plans; provided, however, that the obligations of the Company under this clause (B) shall cease upon Executive becoming eligible to participate in a plan of another employer providing substantially similar group health benefits to Executive and his eligible family members and dependents or upon termination of Executive’s COBRA coverage;
7
(C) if such termination occurs after June 30th in any calendar year, pay Executive a pro-rated Incentive Bonus for the fiscal year during which Executive’s employment is terminated (prorated based on the days elapsed in such fiscal year through the Termination Date);
(D) a lump sum cash payment equal to the Executive’s annual maximum Incentive Bonus Amount, payable on the first payroll date following the first anniversary of the Termination Date (all of the payments in clauses (A) - (D) together, the “Severance Payments”); and
(E) all of the Executive’s options to purchase the common stock of the Company and any awards of restricted common stock received by Executive in each case that were subject to vesting shall vest with respect to that number of shares subject thereto that would have vested during the Severance Period had Executive remained employed by the Company during such period, and such vested options (after taking into account the vesting acceleration) shall remain exercisable through the eighteen (18) month anniversary of the Termination Date (but in no event beyond the original term of the equity award), it being understood that to the extent any options that are intended to be “incentive stock options” for purposes of Section 422 of the Internal Revenue Code are exercised after the three month anniversary of the Termination Date, such options will cease to be treated as “incentive stock options” for purposes of Section 422 of the Internal Revenue Code.
8
9
10
(A) With respect to the Generic Company Products, the development, manufacturing, marketing, distribution or sale, or licensing of any pharmaceutical product in the Territory (1) that is an AB-rated generic equivalent of the same Reference Listed Drug product as a Generic Company Product and for which there are four or fewer AB-rated generic equivalent competitors or (2) that is a direct competitor of any Company Product that is a DESI Product and that utilizes the same active pharmaceutical ingredients as such DESI Product;
(B) With respect to the Brand Company Products, the development, manufacturing, marketing, distribution or sale, or licensing of: (1) any pharmaceutical product in the Territory that is an AB-rated generic equivalent of a Brand Company Product and (2) for which either (I) there are three or fewer AB-rated generic equivalent competitors or (II) AB-rated generic equivalents have been available for commercial sale in the Territory for no more than three years; and
(C)The development, manufacturing, marketing, distribution or sale, or licensing of any product utilizing Corticotropin as the active pharmaceutical ingredient.
11
12
For purposes hereof, “Confidential Information” means and means any and all confidential, proprietary or Trade Secret information of the Company or its controlled affiliates not within the public domain, whether disclosed, directly or indirectly, verbally, in writing (including electronically) or by any other means in tangible or intangible form, including that which is conceived or developed by Executive, applicable to or in any way related to: (i) the present or future business activities, products and services, and customers of the Company or its controlled affiliates; (ii) the research and development of the Company or its controlled affiliates; or (iii) the business of any customers or suppliers of the Company or its controlled affiliates. Such Confidential Information includes the following property or information of the Company or its controlled affiliates, by way of example and without limitation, trade secrets, processes, formulas, data, program documentation, customer lists, pricing information, designs, drawings, algorithms, source code, object code, technology, formulae, models, know-how, improvements, pharmaceutical drug and/or devise technologies, inventions, licenses, techniques, all plans or strategies for marketing, development and pricing, government filings and/or reports, inventions, research, development, schematics, designs, test methods and samples, documents, agreements, business plans, financial statements, profit margins and all information concerning existing or potential clients, suppliers or vendors. Confidential Information of the Company also means all
13
similar information disclosed to the Company by third parties that is subject to confidentiality obligations.
The Company shall not be required to advise Executive specifically of the confidential nature of any such information, nor shall the Company be required to affix a designation of confidentiality to any tangible item, in order to establish and maintain its confidential nature. Notwithstanding the preceding to the contrary, Confidential Information shall not include general industry information or information that is publicly available or readily discernable from publicly available products or literature; information that the Executive lawfully acquires from a source other than the Company or its controlled Affiliates or any customer or supplier of the Company or any of its controlled Affiliates (provided that such source is not bound by a confidentiality agreement with the Company or any of its controlled Affiliates); information that is required to be disclosed pursuant to any law, regulation, rule of any governmental body or authority, or stock exchange, or court order; or information that reflects Executive’s own skills, knowledge, know-how and experience gained prior to employment or service and outside of any connection to or relationship with the Company or any of its controlled Affiliates, or the predecessors of any such entities.
For purposes hereof, the term “Trade Secret” shall have the meaning given in the Delaware enactment of the Uniform Trade Secrets Act, and shall include, without limitation, the whole or any portion or phase of any scientific or technical information, design, process, formula, concept, data organization, manual, other system documentation, or any improvement of any thereof, in any case that is valuable and secret (in the sense that it is not generally known to the Company’s competitors).
Notwithstanding the foregoing, the U.S. Defend Trade Secrets Act of 2016 (“DTSA”) provides that an individual shall not be held criminally or civilly liable under any federal or state trade secret law for the disclosure of a trade secret that is made (i) in confidence to a federal, state, or local government official, either directly or indirectly, or to an attorney; and (ii) solely for the purpose of reporting or investigating a suspected violation of law; or (iii) in a complaint or other document filed in a lawsuit or other proceeding, if such filing is made under seal. In addition, DTSA provides that an individual who files a lawsuit for retaliation by an employer for reporting a suspected violation of law may disclose the trade secret to the attorney of the individual and use the trade secret information in the court proceeding, if the individual (A) files any document containing the trade secret under seal; and (B) does not disclose the trade secret, except pursuant to court order.
14
15
16
(A) one Person (or more than one Person acting as a group) acquires (or has acquired during the 12-month period ending on the date of the most recent acquisition by such Person or group) assets of the Company (including its subsidiaries) that have a total gross fair market value equal to more than fifty percent (50%) of the total gross fair market value of all of the assets of the Company immediately before such acquisition or acquisitions, or
(B) one Person, or more than one Person acting as a group, acquires ownership of equity securities of the Company (including by way of merger, consolidation or otherwise) that, together with all equity securities of the Company previously held by such Person or group, constitutes more than fifty percent (50%) of the total fair market value or total voting power of equity securities of the Company.
Notwithstanding the foregoing, a Change in Control shall not include (1) any transaction effected for reincorporation purposes or (2) any transaction that does not constitute a change of ownership of the Company or a substantial portion of its assets within the meaning of Treasury Regulation Section 1.409A-3(i)(5)(v) or (vii).
17
(A)(1) a Termination of Employment by the Company has occurred for any reason other than for Good Cause or (2) a Termination of Employment by Executive for Good Reason has occurred; and
(B)A Change in Control has been consummated; and
(C)the Termination of Employment, has occurred either: (1) within the period beginning on the date of the consummation of the Change in Control and ending on the last day of the twenty-fourth (24th) month following the consummation of a Change in Control; or (2) prior to a Change in Control, if such Termination of Employment was either a condition of the Change in Control or was at the documented request or insistence of a Person which is a party or an Affiliate of a party to the transaction that constitutes or results in to the Change in Control.
18
19
20
To the Company:
ANI Pharmaceuticals, Inc.
210 Main Street West
Baudette, MN 56623
Attn: General Counsel or the Chief Financial Officer
Telephone No.: 218-634-3500
E-Mail: stephen.carey@anipharmaceuticals.com
To Executive:
Ori Gutwerg
at the contact information on file with the Company
Either party may, by notice given in accordance with this Section, specify a new address for notices under this Agreement.
21
[Signature Page Follows]
22
IN WITNESS WHEREOF, the Parties hereto have executed this Executive Employment Agreement as of the day and year first above written.
Company:
ANI PHARMACEUTICALS, INC.
Name: Nikhil Lalwani
Title: Chief Executive Officer
Executive:
/s/ Ori Gutwerg
Ori Gutwerg
Exhibit A: General Release of All Claims
Exhibit B: Indemnification Agreement
[SIGNATURE PAGE TO EXECUTIVE EMPLOYMENT AGREEMENT]
4124-3184-4650.4
EXHIBIT A
GENERAL RELEASE OF ALL CLAIMS
[See Attached]
4124-3184-4650.4
EXHIBIT B
INDEMNIFICATION AGREEMENT
[See Attached]
4124-3184-4650.4
Exhibit 10.28
Execution Version
EXECUTIVE EMPLOYMENT AGREEMENT
THIS EXECUTIVE EMPLOYMENT AGREEMENT (this “Agreement”) between ANI Pharmaceuticals, Inc. (the “Company”) and Chad Gassert (“Executive”) dated as of March 8, 2021. Each of the Company and Executive are sometimes referred to herein individually as a “Party” and together as the “Parties.”
WHEREAS, in connection with the transactions contemplated by that Agreement and Plan of Merger, dated as of March 8, 2021, by and among the Company, Nile Merger Sub LLC, Novitium Pharma, LLC (“Novitium”), Esjay LLC, Chali Properties, LLC, Executive, Muthusamy Shanmugam, Thorappadi Vijayaraj and Shareholders Representative Services LLC as representative of the Company Members (as defined in the Merger Agreement) (the “Merger Agreement”);
WHEREAS, subject to, and effective as of, the consummation of the Merger (as defined in the Merger Agreement), the Company wishes to employ Executive, and Executive wishes to be employed by the Company on the terms and conditions set forth herein.
NOW, THEREFORE, in consideration of the mutual promises, terms, covenants and conditions set forth herein and the performance of each, the Parties hereto, intending to be legally bound, hereby agree as follows:
2
3
4
5
(A) the Company shall continue to pay Executive his then current Base Salary, payable in regular installments in accordance with the Company’s standard payroll procedures but no less frequently than bi-monthly procedures for a period equal to twelve (12) months following the Termination Date (the “Severance Period”);
(B) if Executive elects to receive continuation coverage under the Consolidated Omnibus Budget Reconciliation Act of 1985, as amended (“COBRA”), pay Executive on a monthly basis for the Severance Period, an amount equal to a percentage (based on the portion of the monthly premium costs covered by the Company for Executive's group health, dental and/or vision coverage in effect as of the Termination Date), of Executive’s monthly COBRA premium payment (if any) under the Company’s group health, dental and vision plans; provided, however, that the obligations of the Company under this clause (B) shall cease upon Executive becoming eligible to participate in a plan of another employer providing
6
substantially similar group health benefits to Executive and his eligible family members and dependents or upon termination of Executive’s COBRA coverage;
(C) if such termination occurs after June 30th in any calendar year, pay Executive a pro-rated Incentive Bonus for the fiscal year during which Executive’s employment is terminated (prorated based on the days elapsed in such fiscal year through the Termination Date);
(D) a lump sum cash payment equal to the Executive’s annual maximum Incentive Bonus Amount, payable on the first payroll date following the first anniversary of the Termination Date (all of the payments in clauses (A) - (D) together, the “Severance Payments”); and
(E) all of the Executive’s options to purchase the common stock of the Company and any awards of restricted common stock received by Executive in each case that were subject to vesting shall vest with respect to that number of shares subject thereto that would have vested during the Severance Period had Executive remained employed by the Company during such period, and such vested options (after taking into account the vesting acceleration) shall remain exercisable through the eighteen (18) month anniversary of the Termination Date (but in no event beyond the original term of the equity award), it being understood that to the extent any options that are intended to be “incentive stock options” for purposes of Section 422 of the Internal Revenue Code are exercised after the three month anniversary of the Termination Date, such options will cease to be treated as “incentive stock options” for purposes of Section 422 of the Internal Revenue Code.
7
8
9
10
For purposes hereof, “Confidential Information” means and means any and all confidential, proprietary or Trade Secret information of the Company or its controlled affiliates not within the public domain, whether disclosed, directly or indirectly, verbally, in writing (including electronically) or by any other means in tangible or intangible form, including that which is conceived or developed by Executive, applicable to or in any way related to: (i) the present or future business activities, products and services, and customers of the Company or its controlled affiliates; (ii) the research and development of the Company or its controlled affiliates; or (iii) the business of any customers or suppliers of the Company or its controlled affiliates. Such Confidential Information includes the following property or information of the Company or its controlled affiliates, by way of example and without limitation, trade secrets, processes, formulas, data, program documentation, customer lists, pricing information, designs, drawings, algorithms, source code, object code, technology, formulae, models, know-how, improvements, pharmaceutical drug and/or devise technologies, inventions, licenses, techniques, all plans or strategies for marketing, development and pricing, government filings and/or reports, inventions, research, development, schematics, designs, test methods and samples, documents, agreements, business plans, financial statements, profit margins and all information concerning existing or potential clients, suppliers or vendors. Confidential Information of the Company also means all similar information disclosed to the Company by third parties that is subject to confidentiality obligations.
The Company shall not be required to advise Executive specifically of the confidential nature of any such information, nor shall the Company be required to affix a designation of confidentiality to any tangible item, in order to establish and maintain its confidential nature. Notwithstanding
11
the preceding to the contrary, Confidential Information shall not include general industry information or information that is publicly available or readily discernable from publicly available products or literature; information that the Executive lawfully acquires from a source other than the Company or its controlled affiliates or any customer or supplier of the Company or any of its controlled affiliates (provided that such source is not bound by a confidentiality agreement with the Company or any of its controlled affiliates); information that is required to be disclosed pursuant to any law, regulation, rule of any governmental body or authority, or stock exchange, or court order; or information that reflects Executive’s own skills, knowledge, know-how and experience gained prior to employment or service and outside of any connection to or relationship with the Company or any of its controlled affiliates, or the predecessors of any such entities.
For purposes hereof, the term “Trade Secret” shall have the meaning given in the Delaware enactment of the Uniform Trade Secrets Act, and shall include, without limitation, the whole or any portion or phase of any scientific or technical information, design, process, formula, concept, data organization, manual, other system documentation, or any improvement of any thereof, in any case that is valuable and secret (in the sense that it is not generally known to the Company’s competitors).
Notwithstanding the foregoing, the U.S. Defend Trade Secrets Act of 2016 (“DTSA”) provides that an individual shall not be held criminally or civilly liable under any federal or state trade secret law for the disclosure of a trade secret that is made (i) in confidence to a federal, state, or local government official, either directly or indirectly, or to an attorney; and (ii) solely for the purpose of reporting or investigating a suspected violation of law; or (iii) in a complaint or other document filed in a lawsuit or other proceeding, if such filing is made under seal. In addition, DTSA provides that an individual who files a lawsuit for retaliation by an employer for reporting a suspected violation of law may disclose the trade secret to the attorney of the individual and use the trade secret information in the court proceeding, if the individual (A) files any document containing the trade secret under seal; and (B) does not disclose the trade secret, except pursuant to court order.
12
13
14
(A) one Person (or more than one Person acting as a group) acquires (or has acquired during the 12-month period ending on the date of the most recent acquisition by such Person or group) assets of the Company (including its subsidiaries) that have a total gross fair market value equal to more than fifty percent (50%) of the total gross fair market value of all of the assets of the Company immediately before such acquisition or acquisitions, or
(B) one Person, or more than one Person acting as a group, acquires ownership of equity securities of the Company (including by way of merger, consolidation or otherwise) that, together with all equity securities of the Company previously held by such Person or group, constitutes more than fifty percent (50%) of the total fair market value or total voting power of equity securities of the Company.
Notwithstanding the foregoing, a Change in Control shall not include (1) any transaction effected for reincorporation purposes or (2) any transaction that does not constitute a change of ownership of the Company or a substantial portion of its assets within the meaning of Treasury Regulation Section 1.409A-3(i)(5)(v) or (vii).
(A)(1) a Termination of Employment by the Company has occurred for any reason other than for Good Cause or (2) a Termination of Employment by Executive for Good Reason has occurred; and
(B)A Change in Control has been consummated; and
(C)the Termination of Employment, has occurred either: (1) within the period beginning on the date of the consummation of the Change in Control and ending on the last day of the twenty-fourth (24th) month
15
following the consummation of a Change in Control; or (2) prior to a Change in Control, if such Termination of Employment was either a condition of the Change in Control or was at the documented request or insistence of a Person which is a party or an Affiliate of a party to the transaction that constitutes or results in to the Change in Control.
16
17
18
To the Company:
ANI Pharmaceuticals, Inc.
210 Main Street West
Baudette, MN 56623
Attn: General Counsel or the Chief Financial Officer
Telephone No.: 218-634-3500
E-Mail: stephen.carey@anipharmaceuticals.com
To Executive:
Chad Gassert
at the contact information on file with the Company
Either Party may, by notice given in accordance with this Section 16, specify a new address for notices under this Agreement.
19
20
[Signature Page Follows]
21
IN WITNESS WHEREOF, the Parties hereto have executed this Executive Employment Agreement as of the day and year first above written.
Company:
ANI PHARMACEUTICALS, INC.
By: /s/ Nikhil Lalwani
Name: Nikhil Lalwani
Title: Chief Executive Officer
Executive:
/s/ Chad Gassert
Chad Gassert
Appendix A: Outside Business Interests and Engagements
Exhibit A: General Release of All Claims
Exhibit B: Indemnification Agreement
[SIGNATURE PAGE TO EXECUTIVE EMPLOYMENT AGREEMENT]
100169106_4
APPENDIX A
OUTSIDE BUSINESS INTERESTS AND ENGAGEMENTS
Scitus Pharma Services Pvt. Ltd (10%) - Clinical research organization that conducts BA/BE studies for Novitium Pharma and other generic pharmaceutical companies. The facility is located in Chennai, Tamil Nadu, India
- 23 -
EXHIBIT A
GENERAL RELEASE OF ALL CLAIMS
This General Release of All Claims (the “Release”) between ANI Pharmaceuticals, Inc. (the “Company”) and [Executive] (referred to hereinafter as “you” or the “Executive”) shall be effective as of the Effective Date (as defined below). Each of the Company and Executive are sometimes referred to herein individually as a “Party” and together as the “Parties.”
You covenant not to sue the Released Parties for any of the claims released above, agree
- 24 -
not to participate in any class, collective, representative, or group action that may include any of the claims released above, and will affirmatively opt out of any such class, collective, representative or group action. Further, you agree not to participate in, seek to recover in, or assist in any litigation or investigation by other persons or entities against the Released Parties with respect to matters related to the Company, except as required by law. Your release covers only those claims that arose prior to the execution of this Release. Execution of this Release does not bar any claim for breach of this Release. Additionally, nothing in this Release precludes you from participating in any investigation or proceeding before any federal or state agency or governmental body. However, while you may file a charge and participate in any such proceeding, by signing this Release, you waive any right to bring a lawsuit against the Released Parties with respect to matters related to the Company, and waive any right to any individual monetary recovery in any such proceeding or lawsuit; provided, however, nothing in this Release is intended to impede your ability to report securities law violations to the Securities and Exchange Commission under the Dodd-Frank Act, or to receive a monetary award from a government administered whistleblower-award program. Nothing in this Release waives your right to testify or prohibits you from testifying in an administrative, legislative, or judicial proceeding concerning alleged criminal conduct or alleged sexual harassment when you have been required or requested to attend the proceeding pursuant to a court order, subpoena or written request from an administrative agency or the legislature.
Notwithstanding the foregoing, the waiver and release contained in this Release does not apply to (i) any current or future rights or claims for indemnification you may have pursuant to the Indemnification Agreement entered into between you and the Company effective [DATE] (the “Indemnification Agreement”), or your indemnification rights under any insurance policy in place and the Company’s internal governing documents; (ii) any vested benefits under an employee benefit plan sponsored by the Company to which you are legally entitled; (iii) any claims to enforce your rights under this Release or the surviving provisions of the Employment Agreement; (iv) the right to share in any claim with respect to being a stockholder of the Company; provided that any such recover is predicated on you not individually bringing any claim or cause of action or actively participating in, or assisting in any way, with respect to any stockholder initiated cause of action; or (v) any claim which, as a matter of law, cannot be released by private agreement. If any provision of the waiver and release contained in this Release is found to be unenforceable, it shall not affect the enforceability of the remaining provisions and a court shall enforce all remaining provisions to the full extent permitted by law.
1 NTD: Review period to be determined as it depends on the circumstances around the termination of employment.
- 25 -
“A general release does not extend to claims THAT the creditor OR RELEASING PARTY does not know or suspect to exist in his OR HER favor at the time of executing the release AND THAT, if known by him OR HER, would have materially affected his or HER settlement with the debtor OR RELEASED PARTY.”
You understand and agree that claims or facts in addition to or different from those which are now known or believed by you to exist may hereafter be discovered, but it is your intention to release all claims that you have or may have against the Released Parties, whether known or unknown, suspected or unsuspected.
- 26 -
- 27 -
- 28 -
- 29 -
[Signature Page Follows]
- 30 -
To accept this Release, please sign and date this Release and return it to me. You have until 5:00 p.m. PT on the date that is [21/45] days following your receipt of this Release to review and consider this Release and to provide me with an executed copy thereof, but in no event may you execute this Release prior to your Termination Date. Please indicate your agreement with the above terms by signing below.
Sincerely,
ANI PHARMACEUTICALS, INC.
By:
(Signature)
Name:
Title:
As set forth above in Section 1(b) above, you have up to [21/45] days after receipt of this Release within which to review it and to discuss with an attorney of your own choosing, at your own expense, whether or not you wish to sign it. Furthermore, you have 7 days after you have signed this Release during which time you may revoke this Release. If you wish to revoke this Release, you may do so by delivering a letter of revocation to [NAME], the Company’s [TITLE], no later than the close of business on the 7th day after you sign this Release. Because of the revocation period, if you don’t revoke this Release, you understand that this Release shall not become effective or enforceable until the 8th day after the date you sign this Release.
My agreement with the terms of this Release is signified by my signature below. Furthermore, I acknowledge that I have read and understand this Release and that I sign this release of all claims voluntarily, with full appreciation that at no time in the future may I pursue any of the rights I have waived in this Release.
Signed Dated:
[Executive]
- 31 -
EXHIBIT B
INDEMNIFICATION AGREEMENT
THIS INDEMNIFICATION AGREEMENT, made and executed this [ ] day of [ ], 20__, by and between ANI Pharmaceuticals, Inc., a Delaware corporation (the “Company”), and [ ], an individual resident of the State of [ ] (the “Indemnitee”).
WHEREAS, the Company is aware that, in order to induce highly competent persons to serve the Company as officers, the Company must provide such persons with adequate protection through insurance and indemnification against inordinate risks of claims and actions against them arising out of their service to and activities on behalf of the Company;
WHEREAS, the Company recognizes that the increasing difficulty in obtaining officers’ liability insurance, the increases in the cost of such insurance and the general reductions in the coverage of such insurance have increased the difficulty of attracting and retaining such persons;
WHEREAS, the Board of Directors of the Company has determined that it is essential to the best interests of the Company’s stockholders that the Company act to assure such persons that there will be increased certainty of such protection in the future;
WHEREAS, it is reasonable, prudent and necessary for the Company contractually to obligate itself to indemnify such persons to the fullest extent permitted by applicable law so that they will continue to serve the Company free from undue concern that they will not be so indemnified; and
WHEREAS, the Indemnitee is willing to serve, continue to serve, and take on additional service for or on behalf of the Company or any of its direct or indirect subsidiaries on the condition that he/she be so indemnified.
NOW, THEREFORE, in consideration of the premises and the mutual promises and covenants contained herein, and for other good and valuable consideration, the receipt and sufficiency of which are hereby acknowledged, the Company and the Indemnitee do hereby agree as follows:
1.Service by the Indemnitee. The Indemnitee agrees to serve and/or continue to serve as an officer of the Company faithfully and will discharge his/her duties and responsibilities to the best of his/her ability so long as the Indemnitee is duly appointed in accordance with the provisions of the Amended and Restated Certificate of Incorporation, as amended (the “Certificate”), and
- 32 -
Bylaws, as amended (the “Bylaws”) of the Company and the General Corporation Law of the State of Delaware, as amended (the “DGCL”), or until his/her earlier death, resignation or removal. The Indemnitee may at any time and for any reason resign from such position (subject to any other contractual obligation or other obligation imposed by operation by law), in which event the Company shall have no obligation under this Agreement to continue to retain the Indemnitee in any such position. Nothing in this Agreement shall confer upon the Indemnitee the right to continue in the employ of the Company or affect the right of the Company to terminate the Indemnitee’s employment at any time in the sole discretion of the Company, with or without cause, subject to any contract rights of the Indemnitee created or existing otherwise than under this Agreement.
2.Indemnification. The Company shall indemnify the Indemnitee against all Expenses (as defined below), judgments, fines and amounts paid in settlement actually and reasonably incurred by the Indemnitee as provided in this Agreement to the fullest extent permitted by the Certificate, Bylaws and DGCL or other applicable law in effect on the date of this Agreement and to any greater extent that applicable law may in the future from time to time permit. Without diminishing the scope of the indemnification provided by this Section 2, the rights of indemnification of the Indemnitee provided hereunder shall include, but shall not be limited to, those rights hereinafter set forth, except that no indemnification shall be paid to the Indemnitee:
(a)on account of any action, suit or proceeding in which judgment is rendered against the Indemnitee for disgorgement of profits made from the purchase or sale by the Indemnitee of securities of the Company pursuant to the provisions of Section 16(b) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or similar provisions of any federal, state or local statutory law;
(b)on account of conduct of the Indemnitee which is finally adjudged by a court of competent jurisdiction to have been knowingly fraudulent or to constitute willful misconduct;
(c)in any circumstance where such indemnification is expressly prohibited by applicable law;
(d)with respect to liability for which payment is actually made to the Indemnitee under a valid and collectible insurance policy of the Company or under a valid and enforceable indemnity clause, Bylaw or agreement (other than this Agreement) of the Company, except in respect of any liability in excess of payment under such insurance, clause, Bylaw or agreement;
- 33 -
(e)if a final decision by a court having jurisdiction in the matter shall determine that such indemnification is not lawful (and, in this respect, both the Company and the Indemnitee have been advised that it is the position of the Securities and Exchange Commission that indemnification for liabilities arising under the federal securities laws is against public policy and is, therefore, unenforceable, and that claims for indemnification should be submitted to the appropriate court for adjudication); or
(f)in connection with any action, suit or proceeding by the Indemnitee against the Company or any of its direct or indirect subsidiaries or the directors, officers, employees or other Indemnitees of the Company or any of its direct or indirect subsidiaries, (i) unless such indemnification is expressly required to be made by law, (ii) unless the proceeding was authorized by the Board of Directors of the Company, (iii) unless such indemnification is provided by the Company, in its sole discretion, pursuant to the powers vested in the Company under applicable law, or (iv) except as provided in Sections 11 and 13 hereof.
3.Actions or Proceedings Other Than an Action by or in the Right of the Company. The Indemnitee shall be entitled to the indemnification rights provided in this Section 3 if the Indemnitee was or is a party or witness or is threatened to be a party or witness to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative in nature, other than an action by or in the right of the Company, by reason of the fact that the Indemnitee is or was a director, officer, employee, agent or fiduciary of the Company, or any of its direct or indirect subsidiaries, or is or was serving at the request of the Company, or any of its direct or indirect subsidiaries, as a director, officer, employee, agent or fiduciary of any other entity, including, but not limited to, another corporation, partnership, limited liability company, employee benefit plan, joint venture, trust or other enterprise, or by reason of any act or omission by him/her in such capacity. Pursuant to this Section 3, the Indemnitee shall be indemnified against all Expenses, judgments, penalties (including excise and similar taxes), fines and amounts paid in settlement which were actually and reasonably incurred by the Indemnitee in connection with such action, suit or proceeding (including, but not limited to, the investigation, defense or appeal thereof), if the Indemnitee acted in good faith and in a manner the Indemnitee reasonably believed to be in or not opposed to the best interests of the Company, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his/her conduct was unlawful.
4.Actions by or in the Right of the Company. The Indemnitee shall be entitled to the indemnification rights provided in this Section 4 if the Indemnitee was or is a party or witness or is threatened to be made a party or witness to any threatened, pending or completed action, suit or proceeding brought by or in the right of the Company to procure a judgment in its favor by reason
- 34 -
of the fact that the Indemnitee is or was a director, officer, employee, agent or fiduciary of the Company, or any of its direct or indirect subsidiaries, or is or was serving at the request of the Company, or any of its direct or indirect subsidiaries, as a director, officer, employee, agent or fiduciary of another entity, including, but not limited to, another corporation, partnership, limited liability company, employee benefit plan, joint venture, trust or other enterprise, or by reason of any act or omission by him/her in any such capacity. Pursuant to this Section 4, the Indemnitee shall be indemnified against all Expenses actually and reasonably incurred by him/her in connection with the defense or settlement of such action, suit or proceeding (including, but not limited to the investigation, defense or appeal thereof), if the Indemnitee acted in good faith and in a manner the Indemnitee reasonably believed to be in or not opposed to the best interests of the Company; provided however, that no such indemnification shall be made in respect of any claim, issue, or matter as to which the Indemnitee shall have been adjudged to be liable to the Company, unless and only to the extent that the Court of Chancery of the State of Delaware or the court in which such action, suit or proceeding was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, the Indemnitee is fairly and reasonably entitled to be indemnified against such Expenses actually and reasonably incurred by him/her which such court shall deem proper.
5.Good Faith Definition. For purposes of this Agreement, the Indemnitee shall be deemed to have acted in good faith and in a manner the Indemnitee reasonably believed to be in or not opposed to the best interests of the Company, or, with respect to any criminal action or proceeding to have had no reasonable cause to believe the Indemnitee’s conduct was unlawful, if, among other things, such action was based on (i) the records or books of the account of the Company or other enterprise, including financial statements; (ii) the advice of legal counsel for the Company or other enterprise; or (iii) information or records given in reports made to the Company or other enterprise by an independent certified public accountant or by an appraiser or other expert selected with reasonable care by the Company or other enterprise.
6.Indemnification for Expenses of Successful Party. Notwithstanding the other provisions of this Agreement, to the extent that the Indemnitee has served on behalf of the Company, or any of its direct or indirect subsidiaries, as a witness or other participant in any class action or proceeding, or has been successful, on the merits or otherwise, in defense of any action, suit or proceeding referred to in Section 3 and 4 hereof, or in defense of any claim, issue or matter therein, including, but not limited to, the dismissal of any action without prejudice, the Indemnitee shall be indemnified against all Expenses actually and reasonably incurred by the Indemnitee in connection therewith, regardless of whether or not the Indemnitee has met the applicable standards of Section 3 or 4 and without any determination pursuant to Section 8.
- 35 -
7.Partial Indemnification. If the Indemnitee is entitled under any provision of this Agreement to indemnification by the Company for some or a portion of the Expenses, judgments, fines and amounts paid in settlement actually and reasonably incurred by the Indemnitee in connection with the investigation, defense, appeal or settlement of such suit, action, investigation or proceeding described in Section 3 or 4 hereof, but is not entitled to indemnification for the total amount thereof, the Company shall nevertheless indemnify the Indemnitee for the portion of such Expenses, judgments, penalties, fines and amounts paid in settlement actually and reasonably incurred by the Indemnitee to which the Indemnitee is entitled.
8.Procedure for Determination of Entitlement to Indemnification.
(a)To obtain indemnification under this Agreement, the Indemnitee shall submit to the Company a written request, including documentation and information which is reasonably available to the Indemnitee and is reasonably necessary to determine whether and to what extent the Indemnitee is entitled to indemnification. The Secretary of the Company shall, promptly upon receipt of a request for indemnification, advise the Board of Directors in writing that the Indemnitee has requested indemnification. Any Expenses incurred by the Indemnitee in connection with the Indemnitee’s request for indemnification hereunder shall be borne by the Company. The Company hereby indemnifies and agrees to hold the Indemnitee harmless for any Expenses incurred by the Indemnitee under the immediately preceding sentence irrespective of the outcome of the determination of the Indemnitee’s entitlement to indemnification.
(b)Upon written request by the Indemnitee for indemnification pursuant to Section 3 or 4 hereof, the entitlement of the Indemnitee to indemnification pursuant to the terms of this Agreement shall be determined by the following person or persons, who shall be empowered to make such determination: (i) if a Change in Control (as hereinafter defined) shall have occurred, by Independent Counsel (as hereinafter defined) (unless the Indemnitee shall request in writing that such determination be made by the Board of Directors (or a committee thereof) in the manner provided for in clause (ii) of this Section 8(b)) in a written opinion to the Board of Directors, a copy of which shall be delivered to the Indemnitee; or (ii) if a Change in Control shall not have occurred, (A)(1) by the Board of Directors of the Company, by a majority vote of Disinterested Directors (as hereinafter defined) even though less than a quorum, or (2) by a committee of Disinterested Directors designated by majority vote of Disinterested Directors, even though less than a quorum, or (B) if there are no such Disinterested Directors or, even if there are such Disinterested Directors, if the Board of Directors, by the majority vote of Disinterested Directors, so directs, by Independent Counsel in a written opinion to the Board of Directors, a copy of which shall be delivered to the Indemnitee. Such Independent Counsel shall be selected by the Board of Directors and approved by the Indemnitee. Upon failure of the Board of Directors to so select, or
- 36 -
upon failure of the Indemnitee to so approve, such Independent Counsel shall be selected by the Chancellor of the State of Delaware or such other person as the Chancellor shall designate to make such selection. Such determination of entitlement to indemnification shall be made not later than 45 days after receipt by the Company of a written request for indemnification. If the person making such determination shall determine that the Indemnitee is entitled to indemnification as to part (but not all) of the application for indemnification, such person shall reasonably prorate such part of indemnification among such claims, issues or matters. If it is so determined that the Indemnitee is entitled to indemnification, payment to the Indemnitee shall be made within ten days after such determination.
9.Presumptions and Effect of Certain Proceedings.
(a)In making a determination with respect to entitlement to indemnification, the Indemnitee shall be presumed to be entitled to indemnification hereunder and the Company shall have the burden of proof in the making of any determination contrary to such presumption.
(b)If the Board of Directors, or such other person or persons empowered pursuant to Section 8 to make the determination of whether the Indemnitee is entitled to indemnification, shall have failed to make a determination as to entitlement to indemnification within 45 days after receipt by the Company of such request, the requisite determination of entitlement to indemnification shall be deemed to have been made and the Indemnitee shall be absolutely entitled to such indemnification, absent actual and material fraud in the request for indemnification or a prohibition of indemnification under applicable law. The termination of any action, suit, investigation or proceeding described in Section 3 or 4 hereof by judgment, order, settlement or conviction, or upon a plea of nolo contendere or its equivalent, shall not, of itself: (i) create a presumption that the Indemnitee did not act in good faith and in a manner which he/she reasonably believed to be in or not opposed to the best interests of the Company, and, with respect to any criminal action or proceeding, that the Indemnitee has reasonable cause to believe that the Indemnitee’s conduct was unlawful; or (ii) otherwise adversely affect the rights of the Indemnitee to indemnification, except as may be provided herein.
10.Advancement of Expenses. All reasonable Expenses actually incurred by the Indemnitee in connection with any threatened or pending action, suit or proceeding shall be paid by the Company in advance of the final disposition of such action, suit or proceeding, if so requested by the Indemnitee, within 20 days after the receipt by the Company of a statement or statements from the Indemnitee requesting such advance or advances. The Indemnitee may submit such statements from time to time. The Indemnitee’s entitlement to such Expenses shall include those incurred in connection with any proceeding by the Indemnitee seeking an adjudication or award in arbitration
- 37 -
pursuant to this Agreement. Such statement or statements shall reasonably evidence the Expenses incurred by the Indemnitee in connection therewith and shall include or be accompanied by a written affirmation by the Indemnitee of the Indemnitee’s good faith belief that the Indemnitee has met the standard of conduct necessary for indemnification under this Agreement and an undertaking by or on behalf of the Indemnitee to repay such amount if it is ultimately determined that the Indemnitee is not entitled to be indemnified against such Expenses by the Company pursuant to this Agreement or otherwise. Each written undertaking to pay amounts advanced must be an unlimited general obligation but need not be secured, and shall be accepted without reference to financial ability to make repayment.
11.Remedies of the Indemnitee in Cases of Determination not to Indemnify or to Advance Expenses. In the event that a determination is made that the Indemnitee is not entitled to indemnification hereunder or if the payment has not been timely made following a determination of entitlement to indemnification pursuant to Sections 8 and 9, or if Expenses are not advanced pursuant to Section 10, the Indemnitee shall be entitled to a final adjudication in an appropriate court of the State of Delaware or any other court of competent jurisdiction of the Indemnitee’s entitlement to such indemnification or advance. Alternatively, the Indemnitee may, at the Indemnitee’s option, seek an award in arbitration to be conducted by a single arbitrator pursuant to the rules of the American Arbitration Association, such award to be made within 60 days following the filing of the demand for arbitration. The Company shall not oppose the Indemnitee’s right to seek any such adjudication or award in arbitration or any other claim. Such judicial proceeding or arbitration shall be made de novo, and the Indemnitee shall not be prejudiced by reason of a determination (if so made) that the Indemnitee is not entitled to indemnification. If a determination is made or deemed to have been made pursuant to the terms of Section 8 or Section 9 hereof that the Indemnitee is entitled to indemnification, the Company shall be bound by such determination and shall be precluded from asserting that such determination has not been made or that the procedure by which such determination was made is not valid, binding and enforceable. The Company further agrees to stipulate in any such court or before any such arbitrator that the Company is bound by all the provisions of this Agreement and is precluded from making any assertions to the contrary. If the court or arbitrator shall determine that the Indemnitee is entitled to any indemnification hereunder, the Company shall pay all reasonable Expenses actually incurred by the Indemnitee in connection with such adjudication or award in arbitration (including, but not limited to, any appellate proceedings).
12.Notification and Defense of Claim. Promptly after receipt by the Indemnitee of notice of the commencement of any action, suit or proceeding, the Indemnitee will, if a claim in respect thereof is to be made against the Company under this Agreement, notify the Company in writing of the commencement thereof; but the omission to so notify the Company will not relieve the Company from any liability that it may have to the Indemnitee otherwise than under this
- 38 -
Agreement or otherwise, except to the extent that the Company may suffer material prejudice by reason of such failure. Notwithstanding any other provision of this Agreement, with respect to any such action, suit or proceeding as to which the Indemnitee gives notice to the Company of the commencement thereof:
(a)The Company will be entitled to participate therein at its own expense.
(b)Except as otherwise provided in this Section 12(b), to the extent that it may wish, the Company, jointly with any other indemnifying party similarly notified, shall be entitled to assume the defense thereof with counsel reasonably satisfactory to the Indemnitee. After notice from the Company to the Indemnitee of its election to so assume the defense thereof, the Company shall not be liable to the Indemnitee under this Agreement for any legal or other Expenses subsequently incurred by the Indemnitee in connection with the defense thereof other than reasonable costs of investigation or as otherwise provided below. The Indemnitee shall have the right to employ the Indemnitee’s own counsel in such action or lawsuit, but the fees and Expenses of such counsel incurred after notice from the Company of its assumption of the defense thereof shall be at the expense of the Indemnitee unless (i) the employment of counsel by the Indemnitee has been authorized by the Company, (ii) the Indemnitee shall have reasonably concluded that there may be a conflict of interest between the Company and the Indemnitee in the conduct of the defense of such action and such determination by the Indemnitee shall be supported by an opinion of counsel, which opinion shall be reasonably acceptable to the Company, or (iii) the Company shall not in fact have employed counsel to assume the defense of the action, in each of which cases the fees and Expenses of counsel shall be at the expense of the Company. The Company shall not be entitled to assume the defense of any action, suit or proceeding brought by or on behalf of the Company or as to which the Indemnitee shall have reached the conclusion provided for in clause (ii) above.
(c)The Company shall not be liable to indemnify the Indemnitee under this Agreement for any amounts paid in settlement of any action, suit or proceeding effected without its written consent, which consent shall not be unreasonably withheld. The Company shall not be required to obtain the consent of the Indemnitee to settle any action, suit or proceeding which the Company has undertaken to defend if the Company assumes full and sole responsibility for such settlement and such settlement grants the Indemnitee a complete and unqualified release in respect of any potential liability.
(d)If, at the time of the receipt of a notice of a claim pursuant to this Section 12, the Company has an officer liability insurance in effect, the Company shall give prompt notice of the commencement of such proceeding to the insurers in accordance with the procedures set forth in
- 39 -
the respective policies. The Company shall thereafter take all necessary or desirable action to cause such insurers to pay, on behalf of the Indemnitee, all amounts payable as a result of such proceeding in accordance with the terms of the policies.
13.Other Right to Indemnification. The indemnification and advancement of Expenses provided by this Agreement are cumulative, and not exclusive, and are in addition to any other rights to which the Indemnitee may now or in the future be entitled under any provision of the Bylaws or Certificate of the Company, any vote of stockholders or Disinterested Directors, any provision of law or otherwise. Except as required by applicable law, the Company shall not adopt any amendment to its Bylaws or Certificate the effect of which would be to deny, diminish or encumber the Indemnitee’s right to indemnification under this Agreement.
14.Officer Liability Insurance. The Company shall maintain officers’ liability insurance for so long as the Indemnitee’s services are covered hereunder, provided and to the extent that such insurance is available on a commercially reasonable basis. In the event the Company maintains officers’ liability insurance, the Indemnitee shall be named as an insured in such manner as to provide the Indemnitee the same rights and benefits as are accorded to the most favorably insured of the Company’s officers. However, the Company agrees that the provisions hereof shall remain in effect regardless of whether liability or other insurance coverage is at any time obtained or retained by the Company, except that any payments made to, or on behalf of, the Indemnitee under an insurance policy shall reduce the obligations of the Company hereunder.
15.Spousal Indemnification. The Company will indemnify the Indemnitee’s spouse to whom the Indemnitee is legally married at any time the Indemnitee is covered under the indemnification provided in this Agreement (even if the Indemnitee did not remain married to him or her during the entire period of coverage) against any pending or threatened action, suit, proceeding or investigation for the same period, to the same extent and subject to the same standards, limitations, obligations and conditions under which the Indemnitee is provided indemnification herein, if the Indemnitee’s spouse (or former spouse) becomes involved in a pending or threatened action, suit, proceeding or investigation solely by reason of his or her status as the Indemnitee’s spouse, including, without limitation, any pending or threatened action, suit, proceeding or investigation that seeks damages recoverable from marital community property, jointly-owned property or property purported to have been transferred from the Indemnitee to his/her spouse (or former spouse). The Indemnitee’s spouse or former spouse also may be entitled to advancement of Expenses to the same extent that the Indemnitee is entitled to advancement of Expenses herein. The Company may maintain insurance to cover its obligation hereunder with respect to the Indemnitee’s spouse (or former spouse) or set aside assets in a trust or escrow fund for that purpose.
- 40 -
16.Intent. This Agreement is intended to be broader than any statutory indemnification rights applicable in the State of Delaware and shall be in addition to any other rights the Indemnitee may have under the Company’s Certificate, Bylaws, applicable law or otherwise. To the extent that a change in applicable law (whether by statute or judicial decision) permits greater indemnification by agreement than would be afforded currently under the Company’s Certificate, Bylaws, applicable law or this Agreement, it is the intent of the parties that the Indemnitee enjoy by this Agreement the greater benefits so afforded by such change. In the event of any change in applicable law, statute or rule which narrows the right of a Delaware corporation to indemnify its officer, employee, agent or fiduciary, such change, to the extent not otherwise required by such law, statute or rule to be applied to this Agreement, shall have no effect on this Agreement or the parties’ rights and obligations hereunder.
17.Attorney’s Fees and Other Expenses to Enforce Agreement. In the event that the Indemnitee is subject to or intervenes in any action, suit or proceeding in which the validity or enforceability of this Agreement is at issue or seeks an adjudication or award in arbitration to enforce the Indemnitee’s rights under, or to recover damages for breach of, this Agreement the Indemnitee, if he/she prevails in whole or in part in such action, shall be entitled to recover from the Company and shall be indemnified by the Company against any actual expenses for attorneys’ fees and disbursements reasonably incurred by the Indemnitee.18.Effective Date. The provisions of this Agreement shall cover claims, actions, suits or proceedings whether now pending or hereafter commenced and shall be retroactive to cover acts or omissions or alleged acts or omissions which heretofore have taken place. The Company shall be liable under this Agreement, pursuant to Sections 3 and 4 hereof, for all acts of the Indemnitee while serving as an officer, notwithstanding the termination of the Indemnitee’s service, if such act was performed or omitted to be performed during the term of the Indemnitee’s service to the Company.
19.Duration of Agreement. This Agreement shall survive and continue even though the Indemnitee may have terminated his/her service as an officer, employee, agent or fiduciary of the Company or as a director, officer, employee, agent or fiduciary of any other entity, including, but not limited to another corporation, partnership, limited liability company, employee benefit plan, joint venture, trust or other enterprise or by reason of any act or omission by the Indemnitee in any such capacity. This Agreement shall be binding upon the Company and its successors and assigns, including, without limitation, any corporation or other entity which may have acquired all or substantially all of the Company’s assets or business or into which the Company may be consolidated or merged, and shall inure to the benefit of the Indemnitee and his/her spouse, successors, assigns, heirs, devisees, executors, administrators or other legal representations. The Company shall require any successor or assignee (whether direct or indirect, by purchase, merger, consolidation or otherwise) to all or substantially all of the business and/or assets of the Company, by written agreement in form and substance reasonably satisfactory to the Company and the
- 41 -
Indemnitee, expressly to assume and agree to perform this Agreement in the same manner and to the same extent that the Company would be required to perform if no such succession or assignment had taken place
.20.Disclosure of Payments. Except as expressly required by any Federal or state securities laws or other Federal or state law, neither party shall disclose any payments under this Agreement unless prior approval of the other party is obtained
.21.Severability. If any provision or provisions of this Agreement shall be held invalid, illegal or unenforceable for any reason whatsoever, (a) the validity, legality and enforceability of the remaining provisions of this Agreement (including, but not limited to, all portions of any Sections of this Agreement containing any such provision held to be invalid, illegal or unenforceable) shall not in any way be affected or impaired thereby and (b) to the fullest extent possible, the provisions of this Agreement (including, but not limited to, all portions of any paragraph of this Agreement containing any such provision held to be invalid, illegal or unenforceable, that are not themselves invalid, illegal or unenforceable) shall be construed so as to give effect to the intent manifest by the provision held invalid, illegal or unenforceable.
22.Counterparts. This Agreement may be executed by one or more counterparts, each of which shall for all purposes be deemed to be an original but all of which together shall constitute one and the same Agreement. Only one such counterpart signed by the party against whom enforceability is sought shall be required to be produced to evidence the existence of this Agreement.
23.Captions. The captions and headings used in this Agreement are inserted for convenience only and shall not be deemed to constitute part of this Agreement or to affect the construction thereof
.24.Definitions. For purposes of this Agreement:
(a)“Change in Control” shall mean the occurrence of any one of the following:
(i)the sale, lease, exchange or other transfer, directly or indirectly, of substantially all of the assets of the Company (in one transaction or in a series of related transactions) to a person or entity that is not controlled by the Company;
- 42 -
(ii)the approval by the stockholders of the Company of any plan or proposal for the liquidation or dissolution of the Company;
(iii)any person becomes after the effective date of this Agreement the “beneficial owner” (as defined in Rule 13d-3 under the Exchange Act), directly or indirectly, of (A) 20% or more, but not 50% or more, of the combined voting power of the Company’s outstanding securities ordinarily having the right to vote at elections of directors, unless the transaction resulting in such ownership has been approved in advance by the Continuity Directors, or (B) 50% or more of the combined voting power of the Company’s outstanding securities ordinarily having the right to vote at elections of directors (regardless of any approval by the Continuity Directors);
(iv)a merger or consolidation to which the Company is a party if the stockholders of the Company immediately prior to effective date of such merger or consolidation have “beneficial ownership” (as defined in Rule 13d-3 under the Exchange Act), immediately following the effective date of such merger or consolidation, of securities of the surviving corporation representing (A) more than 50%, but less than 80%, of the combined voting power of the surviving corporation’s then outstanding securities ordinarily having the right to vote at elections of directors, unless such merger or consolidation has been approved in advance by the Continuity Directors, or (B) 50% or less of the combined voting power of the surviving corporation’s then outstanding securities ordinarily having the right to vote at elections of directors (regardless of any approval by the Continuity Directors);
(v)the Continuity Directors cease for any reason to constitute at least a majority of the Board; or
(vi)any other change in control of the Company of a nature that would be required to be reported pursuant to Section 13 or 15(d) of the Exchange Act, whether or not the Company is then subject to such reporting requirement.
(b)“Continuity Directors” shall mean any individuals who are members of the Board on the effective date of this Agreement and any individual who subsequently becomes a member of the Board whose election, or nomination for election by the Company’s stockholders, was approved by a vote of at least a majority of the Continuity Directors (either by specific vote or by approval of the Company’s proxy statement in which such individual is named as a nominee for director without objection to such nomination).
- 43 -
(c)“Disinterested Director” shall mean a director of the Company who is not or was not a party to the action, suit, investigation or proceeding in respect of which indemnification is being sought by the Indemnitee.
(d)“Expenses” shall include all attorneys’ fees, retainers, court costs, transcript costs, fees of experts, witness fees, travel expenses, duplicating costs, printing and binding costs, telephone charges, postage, delivery service fees, and all other disbursements or expenses incurred in connection with prosecuting, defending, preparing to prosecute or defend, investigating or being or preparing to be a witness in any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative in nature.
(e)“Independent Counsel” shall mean a law firm or a member of a law firm that neither is presently nor in the past five years has been retained to represent (i) the Company or the Indemnitee in any matter material to either such party or (ii) any other party to the action, suit, investigation or proceeding giving rise to a claim for indemnification hereunder. Notwithstanding the foregoing, the term “Independent Counsel” shall not include any person who, under the applicable standards of professional conduct then prevailing, would have a conflict of interest in representing either the Company or the Indemnitee in an action to determine the Indemnitee’s right to indemnification under this Agreement.
25.Entire Agreement, Modification and Waiver. This Agreement constitutes the entire agreement and understanding of the parties hereto regarding the subject matter hereof, and no supplement, modification or amendment of this Agreement shall be binding unless executed in writing by both parties hereto. No waiver of any of the provisions of this Agreement shall be deemed or shall constitute a waiver of any other provisions hereof (whether or not similar) nor shall such waiver constitute a continuing waiver. No supplement, modification or amendment of this Agreement shall limit or restrict any right of the Indemnitee under this Agreement in respect of any act or omission of the Indemnitee prior to the effective date of such supplement, modification or amendment unless expressly provided therein.
26.Notices. All notices, requests, demands or other communications hereunder shall be in writing and shall be deemed to have been duly given if (a) delivered by hand with receipt acknowledged by the party to whom said notice or other communication shall have been directed, (b) mailed by certified or registered mail, return receipt requested with postage prepaid, on the date shown on the return receipt, (c) sent by a recognized next-day courier service on the first business day following the date of dispatch or (d) delivered by facsimile transmission on the date shown on the facsimile machine report:(
- 44 -
i) If to the Indemnitee to:
[NAME]
at the address on file with the Company
Telephone No.: [INSERT]
E-Mail:
(ii) If to the Company, to:
ANI Pharmaceuticals, Inc.
210 Main Street West
Baudette, Minnesota 56623
Attn: Chief Executive Officer
Fax: [(302) 482-8645]
or to such other address as may be furnished to the Indemnitee by the Company or to the Company by the Indemnitee, as the case may be.
27.Governing Law. The parties hereto agree that this Agreement shall be governed by, and construed and enforced in accordance with, the laws of the State of Delaware, applied without giving effect to any conflicts-of-law principles.[Signature Page Follows]
- 45 -
IN WITNESS WHEREOF, the parties hereto have executed this Agreement on the day and year first above written.
ANI PHARMACEUTICALS, INC.
Name:
Title:
INDEMNITEE:
Exhibit 21
ANI PHARMACUTICALS, INC.
The following is a list of subsidiaries of ANI Pharmaceuticals, Inc., omitting subsidiaries which, considered in the aggregate as a single subsidiary, would not constitute a significant subsidiary, as of December 31, 2021:
Name |
| Jurisdiction of Incorporation or Organization |
ANIP Acquisition Company |
| Delaware |
ANI Pharmaceuticals Canada Inc. |
| Canada |
Novitium Pharma, LLC | | Delaware |
New Castle Pharma LLC | | Delaware |
Novitium Labs Private Limited | | Delaware |
New Castle Pharma Real Estate LLC | | Delaware |
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in the Registration Statements of ANI Pharmaceuticals, Inc. on Form S-3 (Nos. 333-239771 and 333-261731) and on Form S-8 (Nos. 333-196518, 333-214416, 333-218120, 333-250892 and 333-260662) of our reports dated March 15, 2022, on our audits of the consolidated financial statements as of December 31, 2021 and 2020 and for each of the years in the three-year period ended December 31, 2021, and the effectiveness of ANI Pharmaceuticals, Inc. and Subsidiaries’ internal control over financial reporting as of December 31, 2021, which reports are included in this Annual Report on Form 10-K to be filed on or about March 15, 2022.
/s/ EisnerAmper LLP
EISNERAMPER LLP
Philadelphia, Pennsylvania
March 15, 2022
Exhibit 31.1
CERTIFICATION OF CHIEF EXECUTIVE OFFICER PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Nikhil Lalwani, certify that:
1. | I have reviewed this Annual Report on Form 10-K of ANI Pharmaceuticals, Inc.; |
2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a–15(e) and 15d–15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a–15(f) and 15d–15(f)) for the registrant and have: |
(a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
(b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
(c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
(d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
(a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
(b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Date: | March 15, 2022 | /s/ Nikhil Lalwani |
| Nikhil Lalwani | |
| President and Chief Executive Officer | |
Exhibit 31.2
CERTIFICATION OF CHIEF FINANCIAL OFFICER PURSUANT TO
SECTION 302 OF THE SARBANES-OXLEY ACT OF 2002
I, Stephen P. Carey, certify that:
1. | I have reviewed this Annual Report on Form 10-K of ANI Pharmaceuticals, Inc.; |
2. | Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report; |
3. | Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report; |
4. | The registrant’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a–15(e) and 15d–15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a–15(f) and 15d–15(f)) for the registrant and have: |
(a) | Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared; |
(b) | Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles; |
(c) | Evaluated the effectiveness of the registrant’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and |
(d) | Disclosed in this report any change in the registrant’s internal control over financial reporting that occurred during the registrant’s most recent fiscal quarter (the registrant’s fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant’s internal control over financial reporting; and |
5. | The registrant’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant’s auditors and the audit committee of the registrant’s board of directors (or persons performing the equivalent functions): |
(a) | All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant’s ability to record, process, summarize and report financial information; and |
(b) | Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant’s internal control over financial reporting. |
Date: | March 15, 2022 | /s/ Stephen P. Carey |
| Stephen P. Carey | |
| Senior Vice President, Finance and Chief Financial Officer | |
Exhibit 32.1
CERTIFICATION
PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED
PURSUANT TO SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report on Form 10-K of ANI Pharmaceuticals, Inc. (the “Company”) for the year ended December 31, 2021 (the “Report”) as filed with the Securities and Exchange Commission on the date hereof, the undersigned Chief Executive Officer and Chief Financial Officer of the Company hereby certify that, to such officer’s knowledge:
(1) the Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) the information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
This certification is provided solely pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
Dated: March 15, 2022 | /s/ Nikhil Lalwani |
| Nikhil Lalwani |
| President and |
| Chief Executive Officer |
| (principal executive officer) |
Dated: March 15, 2022 | /s/ Stephen P. Carey |
| Stephen P. Carey |
| Senior Vice President, Finance and |
| Chief Financial Officer |
| (principal financial and accounting officer) |
A signed original of this written statement required by Section 906 has been provided to the Company and will be retained by the Company and furnished to the Securities and Exchange Commission or its staff upon request.